Scientists Discover New Treatment Strategy for Xtandi-Resistant Prostate Cancer
 |
| Analysis of transcriptomic data from a cohort of CRPC patients24 categorized based on high and low PRKCI expression revealed the upregulation of the histone methyltransferase EZH2 in PRKCI-low tumors along with increased levels of neuroendocrine markers such as chromogranin A (CHGA) or synaptophysin (SYP), and the reduced expression of AR and AR-dependent genes, including KLK2 and KLK3 |
Scientists Discover New Treatment Strategy for Xtandi-Resistant Prostate Cancer
- Scientists
at Weill Cornell Medicine have uncovered a promising new approach to
treat prostate cancer that has become resistant to enzalutamide, a
common hormone therapy. The research, published in Nature
Communications, reveals how targeting protein production in cancer cells
could help overcome treatment resistance.
The team found that prostate cancer cells lacking a protein called PKCλ/ι become resistant to enzalutamide but are uniquely vulnerable to drugs that block protein synthesis. When researchers combined enzalutamide (Xtandi) with drugs that inhibit protein production, like tomivosertib (eFT508), they were able to overcome the cancer's resistance and stop tumor growth.
"This study reveals a critical mechanism behind treatment resistance in prostate cancer, suggesting new therapeutic approaches," said Dr. Maria Diaz-Meco, one of the study's lead authors. "By understanding how these cancer cells adapt and survive, we may be able to make them newly vulnerable to targeted treatments."
The findings are particularly significant for patients with metastatic castration-resistant prostate cancer (mCRPC) who have developed resistance to standard hormone therapies. The researchers demonstrated the effectiveness of this combination therapy approach in both laboratory studies and animal models.
The research suggests that measuring levels of PKCλ/ι in prostate cancer patients could help doctors predict who might develop resistance to enzalutamide and identify which patients would benefit most from combination therapy with protein synthesis inhibitors.
"Low levels of PKCλ/ι create a specific vulnerability in cancer cells," explains Dr. Diaz-Meco. "By testing tumor samples for this protein, we could potentially identify patients who should receive combination treatment before resistance develops, rather than waiting until standard therapy fails."
PKCλ/ι levels can be measured through standard tissue biopsy analysis using immunohistochemistry, a common diagnostic technique already available in most clinical pathology laboratories. The researchers found that patients whose tumors had low PKCλ/ι levels were more likely to develop resistance to enzalutamide but showed enhanced sensitivity to the combination treatment approach.
While no clinical trial is currently underway, the research team is in discussions with clinical partners to develop a trial that would test the combination of enzalutamide with protein synthesis inhibitors in prostate cancer patients with low PKCλ/ι levels. The protein synthesis inhibitor eFT508 (tomivosertib) has already been tested in clinical trials for other cancers, potentially streamlining the path to prostate cancer trials.
"The next step is to translate these laboratory findings into a clinical trial," says Dr. Moscat. "We're particularly excited because the drugs we're proposing to use in combination with enzalutamide are already in clinical development, which could accelerate the testing process."
- This
research offers new hope for prostate cancer patients who have limited
treatment options after developing resistance to standard therapies. The
findings could lead to more effective, personalized treatment
approaches for advanced prostate cancer.
SIDEBAR: Behind the Science: Multi-Institution Team Powers Prostate Cancer Breakthrough
- The
groundbreaking prostate cancer research emerged from a broad
collaboration of leading cancer institutions, spearheaded by Weill
Cornell Medicine scientists.
The study's senior authors, Drs. Maria Diaz-Meco and Jorge Moscat, both Homer T. Hirst III Professors of Oncology in Pathology at Weill Cornell Medicine, led a team that included researchers from Dana-Farber Cancer Institute, Harvard Medical School, Fred Hutchinson Cancer Center, and Roswell Park Comprehensive Cancer Center.
"This kind of complex research requires diverse expertise and perspectives," says Dr. Diaz-Meco. "Our success stems from bringing together specialists in molecular biology, pathology, and clinical medicine."
The research received substantial backing from the National Cancer Institute, with multiple grants supporting different aspects of the work. Core facilities at Weill Cornell Medicine, including their genomics, proteomics, and microscopy units, provided critical technical support.
Four early-career scientists - Shankha Chatterjee, Juan Linares, Tania Cid-Diaz, and Angeles Duran - made key contributions to the study's findings. Their work, conducted at Weill Cornell's Sandra and Edward Meyer Cancer Center, helped unravel the complex mechanisms behind treatment resistance.
The project exemplifies modern cancer research's collaborative nature, drawing on resources and expertise from multiple institutions to tackle challenging clinical problems. "When you're dealing with complex diseases like prostate cancer," notes Dr. Moscat, "it takes a village of scientists working together to make real progress."
Key strategies against drug-resistant prostate cancer | ScienceDaily
- Date: November 20, 2024
- Source: Weill Cornell Medicine
- Summary: An enzyme called EZH2 has an unexpected role in driving aggressive tumor growth in treatment-resistant prostate cancers, according to a new study. The results could potentially lead to new therapies for patients with limited options and add to the significant progress the teams have made in understanding how advanced prostate cancer develops resistance to treatments that target androgen receptors.
An enzyme called EZH2 has an unexpected role in driving aggressive tumor growth in treatment-resistant prostate cancers, according to a new study by scientists at Weill Cornell Medicine. The results could potentially lead to new therapies for patients with limited options and add to the significant progress the teams have made in understanding how advanced prostate cancer develops resistance to treatments that target androgen receptors.
Prostate cancer is a leading cause of cancer-related death in men, claiming over 30,000 lives annually in the United States. While most prostate cancers initially respond to androgen receptor-blocking therapies, some tumors evolve into a highly aggressive, treatment-resistant form known as neuroendocrine prostate cancer, which no longer relies on androgen signaling and is therefore difficult to treat. Understanding this transition has become a priority for researchers and clinicians.
The new study, led by Drs. Maria Diaz-Meco and Jorge Moscat, both Homer T. Hirst III Professors of Oncology in Pathology and members of the Sandra and Edward Meyer Cancer Center at Weill Cornell Medicine, and published Nov. 20 in Nature Communications, found that the absence of a protein called PKCλ/ι in prostate cancer cells enables EZH2 to drive aggressive growth, even with androgen receptor inhibitors present. Normally, PKCλ/ι limits EZH2's activity. However, in PKCλ/ι-deficient cells treated with androgen receptor inhibitors, an alternative form of EZH2 is produced that has a different function. Instead of repressing tumor-suppressor genes, this form of EZH2 drives rapid protein production and activates growth factors like TGF-β, fostering an environment around the tumor that promotes cancer progression despite androgen receptor inhibition.
"This study reveals a critical mechanism behind treatment resistance in prostate cancer, suggesting new therapeutic approaches," said Dr. Diaz-Meco. "By understanding EZH2's role in this context, we may be able to re-sensitize tumors to androgen receptor inhibitors or make the cancer newly vulnerable to targeted treatments, such as immunotherapies."
In preclinical studies, the team targeted EZH2's alternative activities to assess potential treatment solutions. They found that inhibiting either protein synthesis or the TGF-β pathway effectively reversed resistance in PKCλ/ι-deficient cancer cells. Blocking EZH2's alternative function restored sensitivity to androgen receptor therapies like enzalutamide. Furthermore, since TGF-β is associated with immune suppression in tumors, inhibiting this pathway could enhance immunotherapy effectiveness, a treatment with limited success against prostate cancer alone.
The researchers noted that the absence of PKCλ/ι creates a unique vulnerability in cancer cells, suggesting that combining EZH2 inhibitors with AR-targeted therapies could significantly inhibit tumor growth. However, they caution that the inhibition of EZH2 in tumors with high levels of PKCl/i can sometimes counteract therapeutic effects, underscoring the need for precisely tailored treatments for patients with reduced PKCl/i levels. Given the complexity of the EZH2 pathway, achieving a careful balance is essential to avoid reversing treatment benefits.
This research lays the groundwork for clinical trials combining androgen receptor inhibitors with EZH2 or TGF-β inhibitors for patients with therapy-resistant prostate cancer characterized by PKCλ/ι deficiency. Targeting these pathways offers hope not only to overcome AR resistance but also to broaden treatment options for this challenging form of cancer.
Dr. Moscat emphasized the collaborative efforts behind this study, building on previous findings about PKCλ/ι's role in cancer progression. The study's co-first authors are postdoctoral researcher Dr. Shankha Chatterjee, instructor Dr. Juan Linares, postdoctoral researcher Dr. Tania Cid-Diaz, and assistant professor of research in pathology and laboratory medicine Dr. Angeles Duran, all members of the Moscat and Diaz-Meco laboratories.
Shankha S. Chatterjee, Juan F. Linares, Tania Cid-Diaz, Angeles Duran, Mohd. Imran K. Khan, Marta Osrodek, Nicholas J. Brady, Miguel Reina-Campos, Antonio Marzio, Varadha Balaji Venkadakrishnan, Martin K. Bakht, Francesca Khani, Juan Miguel Mosquera, Brian D. Robinson, Jenna Moyer, Olivier Elemento, Andrew C. Hsieh, David W. Goodrich, David S. Rickman, Himisha Beltran, Jorge Moscat, Maria T. Diaz-Meco. Increased translation driven by non-canonical EZH2 creates a synthetic vulnerability in enzalutamide-resistant prostate cancer. Nature Communications, 2024; 15 (1) DOI: 10.1038/s41467-024-53874-2
Increased translation driven by non-canonical EZH2 creates a synthetic vulnerability in enzalutamide-resistant prostate cancer
Nature Communications volume 15, Article number: 9755 (2024)
Abstract
Overcoming resistance to therapy is a major challenge in castration-resistant prostate cancer (CRPC). Lineage plasticity towards a neuroendocrine phenotype enables CRPC to adapt and survive targeted therapies. However, the molecular mechanisms of epigenetic reprogramming during this process are still poorly understood. Here we show that the protein kinase PKCλ/ι-mediated phosphorylation of enhancer of zeste homolog 2 (EZH2) regulates its proteasomal degradation and maintains EZH2 as part of the canonical polycomb repressive complex (PRC2). Loss of PKCλ/ι promotes a switch during enzalutamide treatment to a non-canonical EZH2 cistrome that triggers the transcriptional activation of the translational machinery to induce a transforming growth factor β (TGFβ) resistance program. The increased reliance on protein synthesis creates a synthetic vulnerability in PKCλ/ι-deficient CRPC.
Introduction
Acquired resistance to targeted therapies in cancer is a rising unmet clinical need1. Although androgen deprivation in prostate cancer (PCa) has proven effective for its early management, resistance to the increasingly potent androgen receptor (AR) pathway inhibitors, such as enzalutamide (ENZA) or abiraterone, is now the major barrier to improve patient survival2. CRPC often remains dependent on AR signaling3,4. However, it is now well established that nearly 40% of these tumors reduce their AR dependency under therapy pressure by transitioning towards alternative cellular states, a poorly understood phenomenon termed lineage plasticity5. This mechanism involves the acquisition of histological features of small cell carcinoma and neuronal differentiation by adenocarcinoma cells, giving rise to neuroendocrine prostate cancer (NEPC), which is also endowed with metastasizing potential4,6,7. Targeting these highly aggressive and therapy-resistant tumor cell lineages is becoming a therapeutic priority for PCa and many other cancers8,9. Recent evidence has identified potential drivers of lineage plasticity in PCa, including concurrent loss of TP53 and RB110, and the aberrant expression of several transcription factors, such as the upregulation of N-MYC11, SOX212, BRN213, FOXA214 and the downregulation of REST15. However, the precise signaling mechanisms controlling these processes are largely unknown and constitute a major gap in the field.
Previous findings from our laboratory demonstrated that the loss of the kinase PKCλ/ι resulted in the metabolic reprogramming of PCa cells to undergo cell plasticity towards NEPC differentiation16. Under these conditions, we found that the activation of mTORC1 led to the upregulation of the serine metabolism, which fuels the methionine salvage pathway to produce S-adenosyl-methionine (SAM), the obligate donor of methyl groups in several key cellular events, including the methylation of DNA and histones; both are critical steps in the regulation of chromatin accessibility during transcriptional activation and repression16. We demonstrated that increased levels of SAM sustained the epigenetic changes necessary for lineage plasticity in PCa16,17. In this regard, our previous studies demonstrated the contribution of DNA methylation to the adenocarcinoma-to-NEPC transition and the acquisition of ENZA resistance in PKCλ/ι-deficient PCa cells16. These results highlighted a previously unappreciated role of PKCλ/ι in generating a metabolically permissive cellular state conducive to epigenetic regulation of cell differentiation.
However, a more comprehensive understanding of the epigenetic control of gene expression in this lineage switch must also consider PRC2 and its catalytic subunit, EZH2. The canonical role of EZH2 is to catalyze the methylation of H3K27 to generate H3K27me3, which marks chromatin repression and reduces its transcriptional accessibility18,19. EZH2 is upregulated in human NEPC samples and mouse models driven by N-Myc overexpression or in response to RB1 deficiency in the context of TP53 or PTEN loss4,10,11. Germane to this phenotype and of great functional relevance, pharmacological inhibition of EZH2 was shown to revert the NEPC stage of the triple Rb1/Trp53/Pten mutant cells, concomitantly with the restoration of AR levels and the subsequent enhanced sensitivity to ENZA10. Studies in human NEPC patient-derived organoids revealed their vulnerability to EZH2 inhibition20, which correlated with the loss of NE features, supporting a role for EZH2 in lineage plasticity in CRPC.
Furthermore, evidence also exists of a non-canonical EZH2 “solo” mechanism of action independent of the PRC2 complex and paradoxically associated with co-occupancy of EZH2 with histone activating marks such as H3K27ac and H3K4me3, an indication of an open chromatin and active gene expression21,22,23. This compelling, although still fragmentary, evidence supports a potentially critical role of EZH2 in PCa lineage plasticity but also highlights the complexity of this process. Thus, it is imperative to better understand the mechanism whereby EZH2 is upregulated during this lineage switch, as well as the precise steps whereby EZH2 reprograms the chromatin to impact the gene expression pathways that govern the adenocarcinoma to NEPC transition and resistance to ENZA treatment. This knowledge is key to identifying vulnerabilities to be exploited to treat this type of highly aggressive and therapy-resistant tumor.
Here, we show that PKCλ/ι controls EZH2 proteasome-mediated degradation via direct phosphorylation and maintains EZH2 as part of the canonical PRC2 repressive complex. Consistently, we demonstrate that PKCλ/ι loss in ENZA-treated PCa cells promotes a switch in EZH2 function from the canonical to a non-canonical mechanism of epigenetic reprogramming, resulting in Yin-Yang 1 (YY1)-driven transcriptional activation of protein translation. Our data show that inhibiting EZH2, YY1, or protein synthesis renders PKCλ/ι-deficient PCa cells hypersensitive to growth inhibition by ENZA, identifying the PKCλ/ι-EZH2:YY1 axis as a synthetic vulnerability.
Results
EZH2 expression is upregulated in PKCλ/ι-deficient prostate cancer
Analysis of transcriptomic data from a cohort of CRPC patients24 categorized based on high and low PRKCI expression revealed the upregulation of the histone methyltransferase EZH2 in PRKCI-low tumors along with increased levels of neuroendocrine markers such as chromogranin A (CHGA) or synaptophysin (SYP), and the reduced expression of AR and AR-dependent genes, including KLK2 and KLK3 (Fig. 1a and Supplementary Fig. 1a–c). These changes are common characteristics of NEPC tumors and are consistent with our published data on the induction of NEPC features in PKCλ/ι-deficient cells16. Furthermore, an inverse correlation between EZH2 and PKCλ/ι levels was also observed in the neuroendocrine regions in prostate tumors from the TRAMP+ and Ptenf/fRb1f/fMYCN+PbCre+ NEPC mouse models25, as shown by double immunofluorescence analysis (Fig. 1b, c and Supplementary Fig. 1d, e). To investigate this observation further, we extended our analysis to a human tissue microarray (TMA) comprising 177 samples from surgically resected CRPC in which we examined EZH2 and PKCλ/ι protein levels. After stratifying patients into high and low PKCλ/ι expression categories, we observed a significant enrichment in nuclear EZH2 staining in tumors with low PKCλ/ι expression compared to those of the high PKCλ/ι category (Fig. 1d, e). This increase in EZH2 was detected in nearly all the NEPC samples and, to a lesser extent, also in benign and CRPC adenocarcinomas (Supplementary Fig. 1f). The analysis of an additional cohort of human primary prostate tumors revealed a similar correlation between increased EZH2 nuclear staining and low PKCλ/ι levels (Supplementary Fig. 1g, h). Likewise, human NEPC patient-derived organoids26 (PDOs) with higher levels of NE markers (WCM1078 and WCM154) exhibited an inverse correlation between EZH2 and PKCλ/ι levels (Fig. 1f, g). Furthermore, the analysis of tumors from Ptenf/fPrkcif/fPbCre+ mice as compared to Ptenf/fPbCre+ controls demonstrated a cause-effect link between the genetic inactivation of PKCλ/ι and increased EZH2 levels (Fig. 1h). To investigate the potential cell-autonomous regulation of EZH2 by PKCλ/ι, we genetically deleted PKCλ/ι in LNCaP and C4-2B prostate epithelial cancer cell lines (sgPRKCI) by CRISPR/Cas9. Both sgPRKCI cell lines showed higher EZH2 levels than sgC controls, as determined by immunoblotting and immunofluorescence (Fig. 1i, j and Supplementary Fig. 1i, j). These observations were further confirmed in a set of mouse organoid lines derived from Ptenf/f, Ptenf/fPrkcif/f, Ptenf/fRb1f/f, and Ptenf/fRb1f/fPrkcif/f mice, in which the different alleles were inactivated by ex vivo infection with Cre-expressing adenoviruses (Fig. 1k, l). Notably, the loss of PKCλ/ι does not influence EZH2 at the transcriptional level, nor does it affect the protein levels of other PRC2 complex components. (Supplementary Fig. 1k, l). These results demonstrate a cell-autonomous role of PKCλ/ι in the regulation of EZH2 levels.
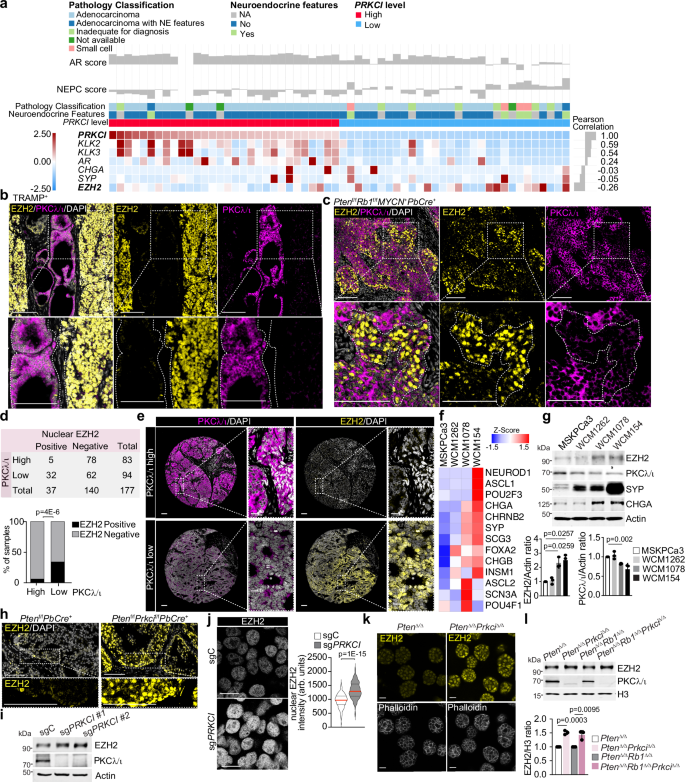
a Heatmap of RNAseq data from the International SU2C/PCF Dream Team metastatic CRPC dataset24 with the inclusion of only the top PRKCI-low (n = 30) and PRKCI-high (n = 30) samples shown. b Representative images of immunofluorescence staining for EZH2, PKCλ/ι, and DAPI in prostates from TRAMP+ mice (n = 3 mice). Scale bars 100 μm. c Representative images of immunofluorescence staining for EZH2, PKCλ/ι, and DAPI in prostate tumors from Ptenf/fRb1f/fMYCN+PbCre+ mice (n = 3 mice per group). Scale bars, 100 μm. d, e Quantification of nuclear EZH2 staining in CRPC samples (n = 177) from a tissue microarray (WCM TMA). CRPC patients were categorized in high or low PKCλ/ι (d). Representative images of immunofluorescence staining for EZH2, PKCλ/ι, and DAPI in CRPC samples from the TMA (n = 177). Scale bars, 100 μm (e). f Heatmap of RNA expression for neuroendocrine (NE)-related genes of human prostate organoids from GSE18137426. g Immunoblots in human adenocarcinoma organoids (MSKPCa3) and NEPC organoids (WCM1262, WCM1078, and WCM154), and quantification (n = 3 independent experiments). h Representative images of immunofluorescence staining for EZH2, and DAPI in prostate tumors from Ptenf/fPbCre+ and Ptenf/fPrkcif/fPbCre+ mice (n = 3 mice per group). Scale bars, 100 μm. i Immunoblots in sgPRKCI and sgC LNCaP cells (n = 3 independent experiments). j Immunofluorescence staining of EZH2 in sgPRKCI and sgC LNCaP cells and quantification of EZH2 intensity (sgC: n = 181, sgPRKCI: n = 139 cells examined). Scale bars, 20 μm. k Representative images of immunofluorescence staining for EZH2, and Phalloidin in PtenΔ/Δ and PtenΔ/ΔPrkciΔ/Δ prostate organoids (n = 3 biological replicates). Scale bars, 20 μm. l Immunoblots in nuclear fraction from PtenΔ/Δ,PtenΔ/ΔPrkciΔ/Δ, PtenΔ/ΔRb1Δ/Δ, and PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ prostate organoids, and quantification (n = 3 independent experiments). Data shown as mean ± SEM (g, j, l). Pearson correlation of pairwise comparisons with PRKCI (a). Two-tailed Chi-square test (d). Two-tailed unpaired Student’s t-test (g, j, l). Source data are provided as a Source Data file.
PKCλ/ι promotes the ubiquitination and subsequent degradation of EZH2
To unravel the precise molecular mechanisms whereby PKCλ/ι regulates EZH2 abundance, we first determined the effect that the ectopic overexpression of PKCλ/ι has on EZH2 levels. Increasing amounts of PKCλ/ι led to the induced reduction of EZH2 protein levels in a dose-dependent manner (Fig. 2a). Cellular proteins undergo degradation through several pathways, including a lysosome-dependent route via autophagy and a NEDD8-dependent or independent route involving the ubiquitin-proteasome system (UPS)27. To determine which of these mechanisms might account for the reduction in EZH2 by PKCλ/ι expression, LNCaP cells were treated either with the proteasome inhibitor MG132, the autophagosome-lysosome inhibitor Bafilomycin A1 or the NEDD8-activating enzyme inhibitor MLN4924 and cultured in the presence or absence of cycloheximide (CHX) to rule out the confounding effect of protein synthesis. Immunoblot analysis showed that transient expression of PKCλ/ι downregulated EZH2 protein levels, which was more apparent in the presence of CHX (Fig. 2b). Treatment with MG132 (but not with the other two inhibitors) completely blocked the decay of EZH2, suggesting that PKCλ/ι regulates the degradation of EZH2 via a UPS-dependent mechanism (Fig. 2b). Furthermore, EZH2 protein was stabilized by PKCλ/ι deficiency with a significant increase in its half-life compared to control cells in two different cell systems (Fig. 2c and Supplementary Fig. 2a). Since the proteasomal degradation pathway involves conjugation of poly-ubiquitin chains to a selected substrate, we hypothesized a potential involvement of PKCλ/ι in the ubiquitination of EZH2. Indeed, loss of PKCλ/ι resulted in reduced EZH2 ubiquitination in LNCaP cells compared to control (Fig. 2d). To identify the ubiquitin ligase that regulates this process, we immunopurified proteins associated with EZH2 expressed in PKCλ/ι-deficient and control cells, followed by mass spectrometry analysis. Among the EZH2-interacting proteins, we identified several putative ubiquitin ligases (Supplementary Fig. 2b), including RBBP6, a RING finger domain-containing E3 ubiquitin ligase whose interaction with EZH2 was significantly reduced in sgPRKCI cells (Fig. 2e and Supplementary Fig. 2b). Both semi-endogenous and endogenous immunoprecipitation analysis validated the impaired interaction between EZH2 and RBBP6 in PKCλ/ι-deficient cells (Fig. 2f, g). No changes were observed in total levels of RBBP6 in sgPRKCI cells (Supplementary Fig. 2c). Consistently, RBBP6 overexpression led to a reduction in EZH2 levels (Fig. 2h), while downregulation of RBBP6 resulted in increased EZH2 as determined by western blot and immunofluorescence analyses (Fig. 2i, j). The accumulation of EZH2 in siRBBP6 cells closely resembled that observed in PKCλ/ι-deficient cells. No further effect on EZH2 levels was detected when RBBP6 was knocked-down in sgPRKCI cells (Fig. 2k). Furthermore, the downregulation of RBBP6 mimicked PKCλ/ι deficiency in impairing EZH2 ubiquitination and the ensuing increased EZH2 stability (Fig. 2l, m), which supports the notion that PKCλ/ι targets EZH2 for ubiquitination through RBBP6, triggering its proteasome-mediated degradation.
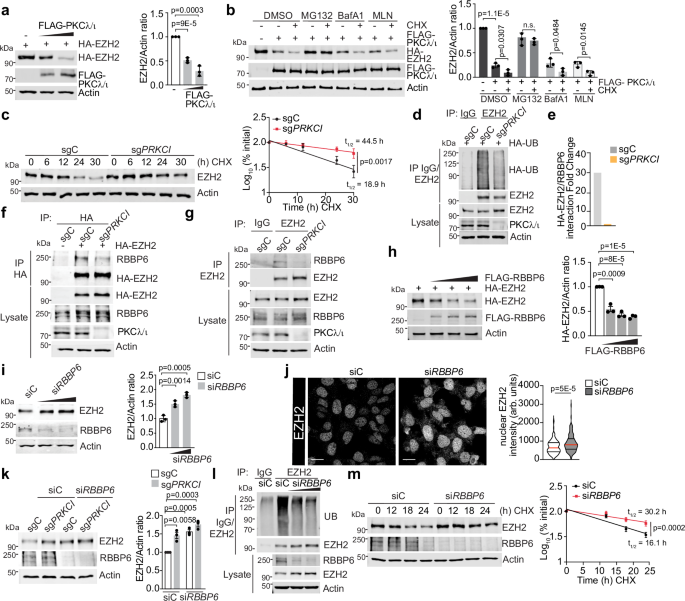
a Immunoblots in HEK293T cells transfected with the indicated plasmids and quantification of HA-EZH2 (n = 3 independent experiments). b Immunoblots in LNCaP cells transfected with the indicated plasmids and treated with cycloheximide (CHX) (50 μg/ml) and MG132 (10 μM), Bafilomycin A1 (100 nM) or MLN4924 (1 μM) for 12 h, and quantification of HA-EZH2 (n = 3 independent experiments). c sgPRKCI and sgC LNCaP cells were incubated with 50 μg/ml of CHX at indicated time points, and quantification of EZH2 (n = 3 independent experiments). d Immunoblotting of EZH2 immunoprecipitates in sgPRKCI and sgC LNCaP cells, transfected with the indicated plasmids (n = 2 independent experiments). e Signal Intensity of EZH2-RBBP6 interaction measured by Mass Spectrometry in sgC and sgPRKCI HEK293T cells transfected with HA-EZH2 (n = 1 sample per condition). f Immunoblotting of HA-tagged immunoprecipitates of sgPRKCI and sgC LNCaP cells, transfected with the indicated plasmids (n = 2 independent experiments). g Immunoblotting of EZH2 immunoprecipitates in sgPRKCI and sgC LNCaP cells (n = 2 independent experiments). h Immunoblots in HEK293T cells, transfected with the indicated plasmids and quantification of EZH2 (n = 3 independent experiments). i Immunoblots in LNCaP cells, transduced with siRNAs and quantification of EZH2 (n = 3 independent experiments). j Immunofluorescence staining of EZH2 in LNCaP cells transduced with siRNAs and quantification of the EZH2 intensity (siC: n = 143, siRBBP6: n = 138 cells examined). Scale bars 10 μm. k Immunoblots in sgPRKCI and sgC LNCaP cells, transduced with the indicated siRNAs and quantification of EZH2 (n = 3 independent experiments). l Immunoblotting of EZH2 immunoprecipitates in LNCaP cells, transduced with the indicated siRNAs (n = 2 independent experiments). m LNCaP cells, transduced with the indicated siRNAs, were treated as in (c), and EZH2 quantification (n = 3 independent experiments). Immunoblot experiments were performed at least two times independently, with similar results. Data shown as mean ± SEM of 3 biological replicates (a, b, c, h, i, k, m). Two-tailed unpaired Student’s t-test (a, b, h, i, j, k). Two-way ANOVA (c, m). Source data are provided as a Source Data file.
Phosphorylation of Ser 380 by PKCλ/ι is critical for EZH2 regulation
An in vitro kinase assay demonstrated that recombinant PKCλ/ι directly phosphorylates EZH2 (Fig. 3a). Therefore, we next employed a dual mass spectrometry analysis approach to map the PKCλ/ι phosphorylation sites in EZH2. Thus, the sites directly phosphorylated in an in vitro assay were compared with those reduced in PKCλ/ι-deficient cells in an in-cell assay (Fig. 3b). This overlap identified five potential sites, namely, S21, S75, S76, S375, and S380 (Fig. 3b). EZH2 S21 was reported as a potential substrate for PI3K/AKT21,28. While we validated that PKCλ/ι phosphorylates that residue using a phospho-specific antibody (Supplementary Fig. 3a), its mutation to alanine did not reduce PKCλ/ι-mediated EZH2 phosphorylation (Supplementary Fig. 3b), indicating a marginal contribution of S21 phosphorylation to the total pool of phosphorylated EZH2. We next generated HA-tagged EZH2 alanine mutants of the other identified sites. Mutation of S380 to alanine (HA-EZH2S380A) resulted in a major reduction of total EZH2 phosphorylation by PKCλ/ι with a minor additional contribution from S75A and S76A (Supplementary Fig. 3c). Notably, S380 was identified in the mass spectrometry database PhosphoSitePlus (https://www.phosphosite.org), confirmed the aPKC consensus phosphorylation motif (Supplementary Fig. 3d), and was predicted as a phosphorylation site for PKCλ/ι using the kinase-specific phosphorylation site prediction GPS 5.0. tool29. Moreover, the double S375/380AA mutant (HA-EZH2S375/380AA) gave a near complete reduction in EZH2 phosphorylation (Fig. 3c and Supplementary Fig. 3c), demonstrating that these two sites are the major EZH2 bona fide direct targets of PKCλ/ι. These residues were conserved across different species (Fig. 3d), highlighting their potential evolutionary significance.
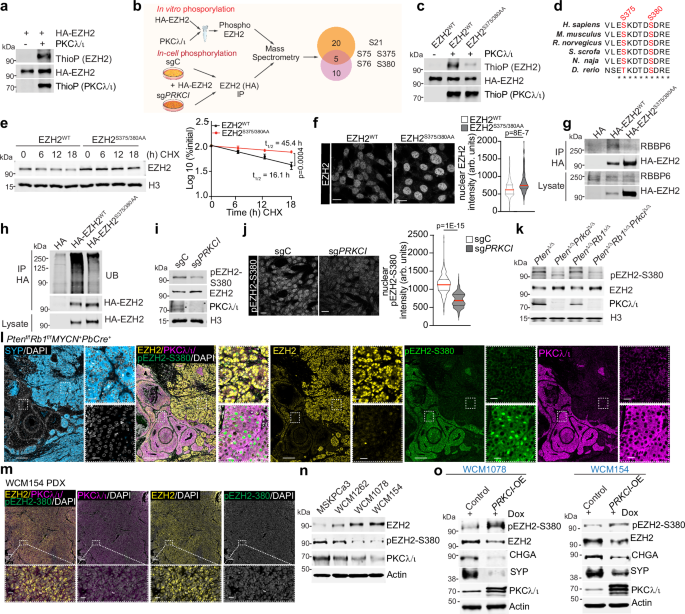
a In vitro phosphorylation of HA-tagged EZH2 by recombinant PKCλ/ι (n = 3 independent experiments). b Identification of EZH2 phosphorylation sites by PKCλ/ι: HA-EZH2, in vitro phosphorylated with recombinant PKCλ/ι, or HA-EZH2 transfected into sgC and sgPRKCI cells were analyzed by MS (n = 1 sample per condition). c In vitro phosphorylation of HA-EZH2WT or EZH2S375/380AA as in (a) (n = 2 independent experiments). d Alignment of the amino acid sequence of human EZH2 (372-383 aa) with orthologs in other species. e EZH2WT or EZH2S375/380AA LNCaP cells were incubated with CHX (50 μg/ml) at indicated time points, and EZH2 levels were quantified (n = 3 independent experiments). f Immunofluorescent staining of EZH2 in EZH2WT or EZH2S375/380AA LNCaP cells and quantification (EZH2WT: n = 91, EZH2S375/380AA: n = 83 cells examined). Scale bars 10 μm. g, h Immunoblots of HA-tagged immunoprecipitates in HEK293T, transfected for the indicated plasmids (n = 2 independent experiments). i Immunoblots in nuclear lysates from sgPRKCI and sgC LNCaP cells (n = 2 independent experiments). j Immunofluorescence staining of pEZH2(S380) in sgPRKCI and sgC LNCaP cells (sgC: n = 459, sgPRKCI: n = 270 cells examined), and quantification. Scale bars 10 μm. k Immunoblots in PtenΔ/Δ, PtenΔ/ΔPrkciΔ/Δ, PtenΔ/ΔRb1Δ/Δ, and PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ prostate organoids (n = 3 independent experiments). l Immunofluorescence staining for pEZH2(S380), EZH2, PKCλ/ι, SYP and DAPI in prostate tumors from the NEPC model Ptenf/fRb1f/fMYCN+PbCre+ (n = 3 mice per group). Scale bars 200 μm and 20 μm. m Immunofluorescence staining for pEZH2(S380), EZH2, PKCλ/ι, and DAPI in human NEPC PDOs WCM154 (n = 1). Scale bars, 100 μm and 20 μm. n Immunoblots in human adenocarcinoma (MSKPCa3) and NEPC (WCM1262, WCM1078, WCM154) organoids (n = 3 independent experiments). o Immunoblots in control and PRKCI-overexpressed (OE) NEPC PDOs WCM1078 and WCM154 (n = 2 independent experiments). Immunoblot experiments were performed at least two times independently, with similar results. Data shown as mean ± SEM of the biological replicates (e). Two-way ANOVA (e). Two-tailed unpaired Student’s t-test (f, j). Source data are provided as a Source Data file.
To determine the functional importance of these phosphorylation sites, we mutated S375A and S380A at the endogenous EZH2 locus in LNCaP cells using CRISPR/Cas9-mediated editing to create the knock-in EZH2S375/380AA cells. CHX chase experiments demonstrated increased stability and prolonged half-life of the EZH2S375/380AA mutant protein compared to EZH2WT in control cells (Fig. 3e). Similar results were obtained using the HA-tagged mutant EZH2S375/380AA transfected in HEK293T cells (Supplementary Fig. 3e). Consistent with this enhanced stability, EZH2S375/380AA cells exhibited higher levels of nuclear EZH2 staining as compared to EZH2WT cells (Fig. 3f). Furthermore, EZH2S375/380AA displayed a reduced binding to RBBP6, resulting in decreased EZH2-ubiquitination and increased protein levels (Fig. 3g, h). The knock-down of RBBP6 does not influence EZH2 phosphorylation status (Supplementary Fig. 3f). These results demonstrate that PKCλ/ι-mediated phosphorylation of EZH2 at S375 and S380 regulates its degradation by recruiting the ubiquitin ligase RBBP6.
To interrogate the biological significance of these observations, we generated phospho-specific antibodies for these sites. While phospho-S375/380 and phospho-S375 antibodies did not show sufficient quality or specificity, we successfully developed and validated a phospho-S380 antibody (pEZH2-S380), which detects the most relevant site in terms of contribution to the total EZH2 phosphorylation by PKCλ/ι (Supplementary Fig. 3c). This antibody specifically recognized an EZH2 peptide with phosphorylated S380 but failed to detect the corresponding unphosphorylated peptide (Supplementary Fig. 3g). Furthermore, incubation with the phosphorylated peptide successfully competed for antibody binding in immunoblotting analysis of whole LNCaP cell extracts (Supplementary Fig. 3h). Consistently, the p-EZH2-380 antibody also reacted with wild-type EZH2 but not with the EZH2S375/380AA mutant (Supplementary Fig. 3i, j), demonstrating its specificity for phosphorylation at the S380 site. Of functional relevance, PKCλ/ι-deficient LNCaP cells displayed a significant reduction in EZH2 S380 phosphorylation despite increased total EZH2 content (Fig. 3i, j). Consistently, pEZH2-S380 levels were also reduced in PKCλ/ι-deficient mouse prostate organoids (Fig. 3k). Additionally, immunohistochemistry analysis of prostate tumors from the Ptenf/fRb1f/fMYCN+PbCre+ NEPC mouse model showed that pEZH2-S380 staining was highly positive in the adenocarcinoma regions that retained PKCλ/ι expression, concomitant with low EZH2 and the absence of the neuronal marker SYP (Fig. 3l). On the contrary, neuroendocrine regions displayed almost negative pEZH2-S380 staining, low PKCλ/ι expression, and high levels of EZH2 and SYP (Fig. 3l). A similar positive correlation between pEZH2-S380 and PKCλ/ι was also observed in liver NEPC metastases from the TRAMP+ mouse model (Supplementary Fig. 3k). These findings were further validated in a human NEPC patient-derived xenograft (PDX) that showed complete lack of EZH2 S380 phosphorylation and PKCλ/ι expression, concurrent with heightened EZH2 abundance (Fig. 3m). Consistently, human NEPC organoid lines, such as WCM1078 and WCM154, characterized by low PKCλ/ι and high levels of NE markers (Fig. 1f, g), exhibited an inverse correlation between EZH2 and pEZH2-S380 levels (Fig. 3n). In contrast, PRKCI overexpression in these organoids led to a reduction in the expression of NE markers such as SYP and CHGA, an increase in S380 phosphorylation of EZH2, and a decrease in total EZH2 levels along with a corresponding rise of PKCλ/ι (Fig. 3o). Collectively, these results demonstrate that the S380 phosphorylation of EZH2 by PKCλ/ι is physiologically relevant.
EZH2 modulation by PKCλ/ι deficiency creates a synthetic vulnerability in ENZA-treated PCa cells
Previous studies have demonstrated that EZH2 is critical in acquiring ENZA resistance in CRPC cells10. Consequently, inhibiting or depleting EZH2 has emerged as a promising therapeutic strategy to reverse ENZA resistance. We evaluated the viability of sgPRKCI and sgC LNCaP cells treated with different doses of ENZA. Consistent with our previous observations16, PKCλ/ι-deficient cells were less sensitive to growth inhibition by ENZA than control cells (Fig. 4a). Furthermore, EZH2S375/380AA cells were less efficiently inhibited by ENZA than EZH2WT cells, indicating that EZH2 phosphorylation by PKCλ/ι has an important role in the response of PCa cells to ENZA treatment (Fig. 4b). To further establish the role of EZH2 upregulation in the reduced sensitivity of PKCλ/ι-deficient PCa cells to ENZA, we pharmacologically inhibited EZH2 catalytic activity with the GSK126 compound. Remarkably, the growth inhibition effect of the combination of both drugs was more potent in sgPRKCI cells than in control cells (Fig. 4c). That is, in the presence of GSK126, the IC50 for ENZA was significantly reduced by nearly 15-fold in PKCλ/ι-deficient cells (Fig. 4c). Furthermore, we observed synergism between GSK126 and ENZA in PKCλ/ι-deficient cells compared to control cells (Fig. 4d). The synergy between both drugs in PKCλ/ι-deficient cells was also demonstrated in a time course experiment in LNCaP cells, as well as in two genetically defined mouse prostate organoids (Fig. 4e–g). Similar results were obtained with EPZ6438, another pharmacological FDA-approved inhibitor of EZH2 catalytic activity (Fig. 4h, i and Supplementary Fig. 4a, b), as well as with the EZH2 pharmacological degrader MS1943 (Fig. 4j and Supplementary Fig. 4c, d). Importantly, these effects were also detected in PKCλ/ι-deficient mouse prostate organoids, as well as in human prostate cancer organoids with low expression of endogenous PKCλ/ι (Figs. 1g, 4i, k). These results demonstrate that PKCλ/ι deficiency while impairing ENZA effects, creates a synthetic vulnerability to the combined treatment of ENZA with the inhibition of EZH2.
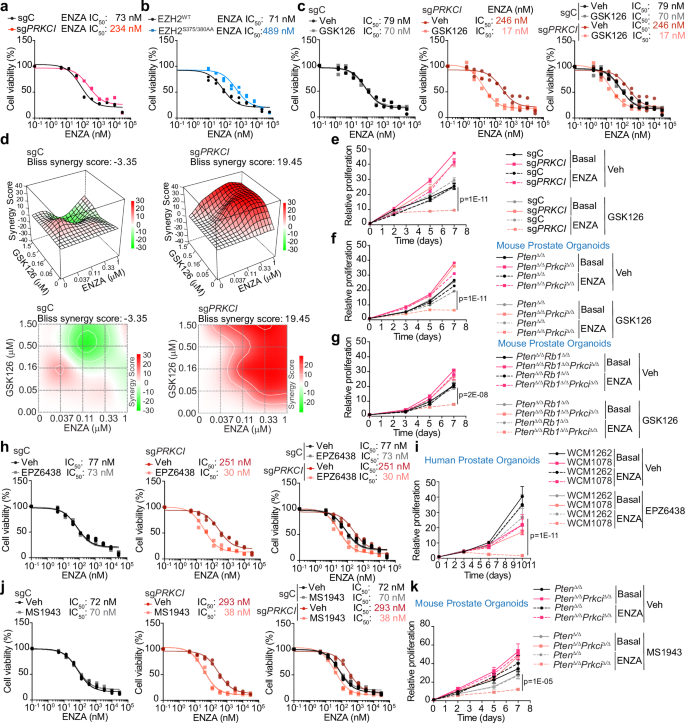
a, b Dose-response curves using CFU assay for 14 days to determine the IC50 of ENZA for sgC and sgPRKCI (a) or EZH2WT, EZH2S375/380AA (b) LNCaP cells. IC50 value as the average of two biological replicates. c Dose-response curves using CFU assay for 14 days to determine the IC50 of ENZA treated with vehicle or 5 μM GSK126 in sgC and sgPRKCI LNCaP cells. IC50 value as the average of two biological replicates. d sgC and sgPRKCI LNCaP cells were treated with ENZA and GSK126 alone or combined and drug synergism was assessed using Bliss Independence method (n = 3 technical replicates). The positive drug synergy is represented as red peaks and the Bliss synergy scores are indicated on the 3-D plots. e Growth curves of sgC and sgPRKCI LNCaP cells treated with 5 μM ENZA and 5 μM GSK126 alone or combined. Representative experiment of two biological replicates. f, g Growth curves of PtenΔ/Δ and PtenΔ/ΔPrkciΔ/Δ (f) and PtenΔ/ΔRb1Δ/Δ and PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ (g) mouse prostate organoids treated with 5 μM ENZA and 10 μM GSK126 alone or combined. Representative experiment of two biological replicates. h Dose-response curves using CFU assay for 14 days to determine the IC50 of ENZA treated with vehicle or 75 nM EPZ6438 in sgC and sgPRKCI LNCaP cells. IC50 value as the average of two biological replicates. i Growth curves of WCM1262 and WCM1078 human prostate organoids treated with 10 μM ENZA and 10 μM EPZ6438 alone or combined. Representative experiment of two biological replicates. j Dose-response curves using CFU assay for 14 days to determine the IC50 of ENZA treated with vehicle or 1 μM MS1943 in sgC and sgPRKCI LNCaP cells. IC50 value is the average of two biological replicates. k Growth curves of PtenΔ/Δ and PtenΔ/ΔPrkciΔ/Δ mouse organoids treated with 5 μM ENZA and 5 μM MS1943 alone or combined. Representative experiment of two biological replicates. Data shown as mean ± SD of technical triplicates (e, f, g, i, k). Bliss synergy scores were calculated using SynergyFinder 2.2 (d). Two-way ANOVA (e, f, g, i, k). Source data are provided as a Source Data file.
PKCλ/ι loss reduces canonical and drives non-canonical EZH2 function
To examine the effect of PKCλ/ι loss on genome-wide EZH2-mediated epigenetic remodeling and gene regulation, we performed cleavage under targets and release using nuclease (CUT&RUN) sequencing studies in LNCaP cells under basal or ENZA-treated conditions using antibodies for EZH2, H3K27me3, or H3K4me3. Under basal conditions, most sites were co-occupied by EZH2 and the repressive H3K27me3 mark and were termed “EZH2 ensemble”, corresponding to the canonical PRC2 complex (Fig. 5a and Supplementary Fig. 5a–c). We also identified a minor subset of EZH2 bound regions that were devoid of H3K27me3 but overlapped with the H3K4me3 activation mark, which we termed as “EZH2 solo” (Fig. 5a and Supplementary Fig. 5a–c). However, under ENZA-treated conditions, there was a major switch in the EZH2 landscape in PKCλ/ι-deficient cells, characterized by a marked gain in the EZH2 solo and a reduction in the EZH2 ensemble peaks (Fig. 5a, b and Supplementary Fig. 5d, e). EZH2 solo occupancy was enhanced in the promoter and intronic regions while reduced in the intergenic regions (Fig. 5c). Moreover, unique EZH2 solo peaks in sgPRKCI were mostly in promoters when compared to those in control cells (Supplementary Fig. 5f), indicating that EZH2 solo binding sites regulate gene expression differently from the canonical PRC2 complex.
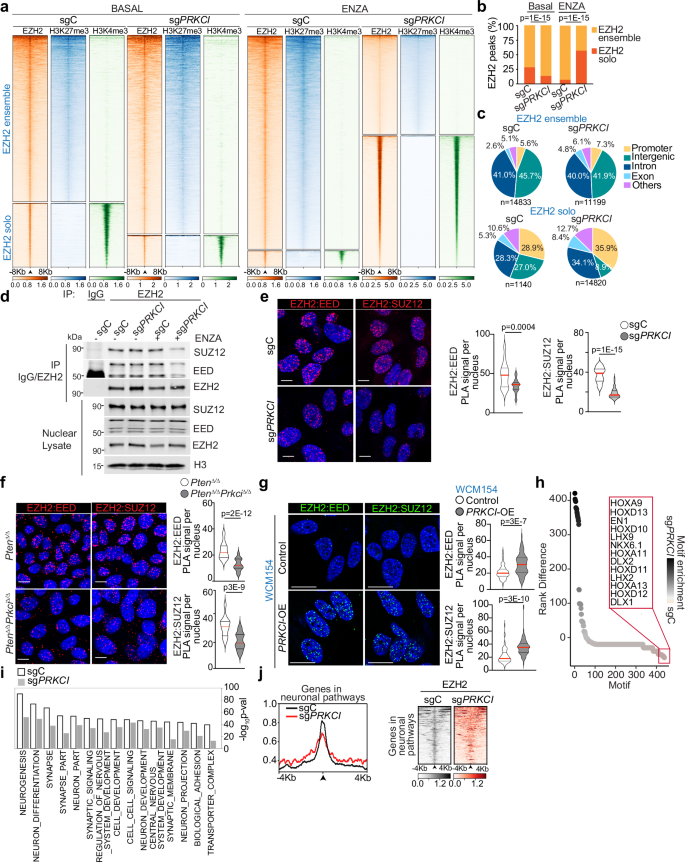
a Heatmaps (CUT&RUN) for EZH2, H3K27me3 and H3K4me3 ± 8 kb from the centers of canonical EZH2+/H3K27me3+ peaks (EZH2 ensemble; top panels) or non-canonical EZH2+/H3K27me-/H3K4me3+ peaks (EZH2 solo; bottom panels) in sgPRKCI and sgC LNCaP cells treated or not with 10 μM ENZA for 72 h (n = 3 biological replicates). b Percentage of EZH2 solo peaks and ensemble peaks found in (a) (n = 3 biological replicates). c Pie-chart plot showing the genomic distribution of peaks for EZH2 ensemble or solo in sgPRKCI and sgC LNCaP cells, treated as in (a) (n = 3 biological replicates). d Immunoblotting of nuclear lysates and EZH2 immunoprecipitates of sgC and sgPRKCI LNCaP cells, treated as in (a) (n = 2 independent experiments). e–g Proximity Ligation Assay (PLA) of EZH2-EED or EZH2-SUZ12 in sgPRKCI (EZH2-EED: n = 25; EZH2-SUZ12: 40 cells examined) and sgC (EZH2-EED: n = 33; EZH2-SUZ12: 40 cells examined) LNCaP cells (e), PtenΔ/Δ (EZH2-EED: n = 39; EZH2-SUZ12: 39 cells examined) and PtenΔ/ΔPrkciΔ/Δ (EZH2-EED: n = 39; EZH2-SUZ12: 34 cells examined) mouse organoids (f), or PRKCI-OE (EZH2-EED: n = 82; EZH2-SUZ12: 75 cells examined) and Control (EZH2-EED: n = 68; EZH2-SUZ12: 75 cells examined) NEPC PD)Os WCM154 (g) treated as in (a), and quantification. Scale bars, 10 μm. h Enrichment of differential transcription factor motifs between EZH2 ensemble peaks (all genomic regions) in sgPRKCI and sgC LNCaP cells, plotted by ranks generated from their associated p values (n = 3 biological replicates). i Top 15 GO pathways from findGO.pl, analysis of genes with unique EZH2 ensemble peaks in sgPRKCI and sgC LNCaP cells treated with ENZA (n = 3 biological replicates). j Averaged signal intensities and Heatmap (CUT&RUN) for EZH2 ± 4 kb from the centers of EZH2 ensemble peaks in genes from neuronal-related pathways (n = 3 biological replicates). Immunoblot experiments were performed at least two times independently with similar results. Data shown as mean ± SEM (e–g). Fisher’s exact test (b). Two-tailed unpaired Student’s t-test (e–g). Source data are provided as a Source Data file.
In keeping with the ensemble-to-solo complex switch in sgPRKCI cells, immunoprecipitation experiments in PKCλ/ι-deficient LNCaP cells demonstrated an impaired interaction between EZH2 and EED or SUZ12, essential subunits of the PRC2 complex, under ENZA-treated conditions (Fig. 5d). Moreover, in situ proximity ligation assays (PLA) in PKCλ/ι-deficient LNCaP cells and mouse prostate organoids confirmed the reduced interaction of EZH2 with EED and revealed an impaired binding of EZH2 to SUZ12, the other core subunit of the PRC2 holoenzyme (Fig. 5e, f). In contrast, PRKCI overexpression in NEPC WCM154 organoids with reduced levels of PKCλ/ι rescued the interaction between EZH2 and EED or SUZ12 (Fig. 5g). Consistent with the role of PKCλ/ι in the PRC2 assembly, the loss of PKCλ/ι resulted in a reduction of H3K27me3 levels in ENZA-treated cells (Supplementary Fig. 5g). Moreover, treatment with other androgen receptor signaling inhibitors such as Apalutamide or Darolutamide, caused similar impairments in the EZH2-EED interaction (Supplementary Fig. 5h) as observed with ENZA treatment in PKCλ/ι-deficient cells. Motif analysis of the reduced ensemble EZH2 peaks in PKCλ/ι-deficient cells revealed enrichment for the binding of multiple transcription factors (TFs) of the Homeobox family associated with neuronal cell fate (EN1, DLX1, NKX family, HOX family, and LHX family) (Fig. 5h and Supplementary Fig. 5i). Interestingly, pathway analysis of EZH2 ensemble unique peaks in sgPRKCI cells was consistent with a decrease in the repression of pathways related to neuronal development (Fig. 5i and Supplementary Fig. 5j), correlating with the significant reduction of EZH2 binding in a subset of genes involved in neuronal pathways (Fig. 5j). These results are in agreement with our previously reported observations that PKCλ/ι loss promotes the emergence of NEPC features in PCa cells and drives prostate neoplasia in vivo16.
PKCλ/ι deficiency drives ENZA resistance through a non-canonical EZH2:YY1 complex
TF motif analysis of EZH2 solo peaks in ENZA-treated PKCλ/ι-deficient cells around all genomic positions revealed significant enrichment in YY1 and factors associated with the ETV and ETS family of genes (Fig. 6a). Subsequent interrogation of the peaks at the transcription start site (TSS) and flanking 2 kb region, again indicated significant enrichment in YY1 (Supplementary Fig. 6a). We next performed CUT&RUN for YY1 in sgC and sgPRKCI LNCaP cells under ENZA-treated conditions and calculated the overlap of YY1 signal with uniquely bound EZH2 solo or ensemble peaks in each condition (Fig. 6b, c and Supplementary Fig. 6b, c). A robust and higher percentage of YY1 overlap was observed in PKCλ/ι-deficient LNCaP cells (51.6%) compared to sgC cells (9.4%) in EZH2 solo regions (Fig. 6c). In contrast, a minimal overlap between EZH2 and YY1 peaks was observed in EZH2 ensemble regions under both conditions (Fig. 6c).
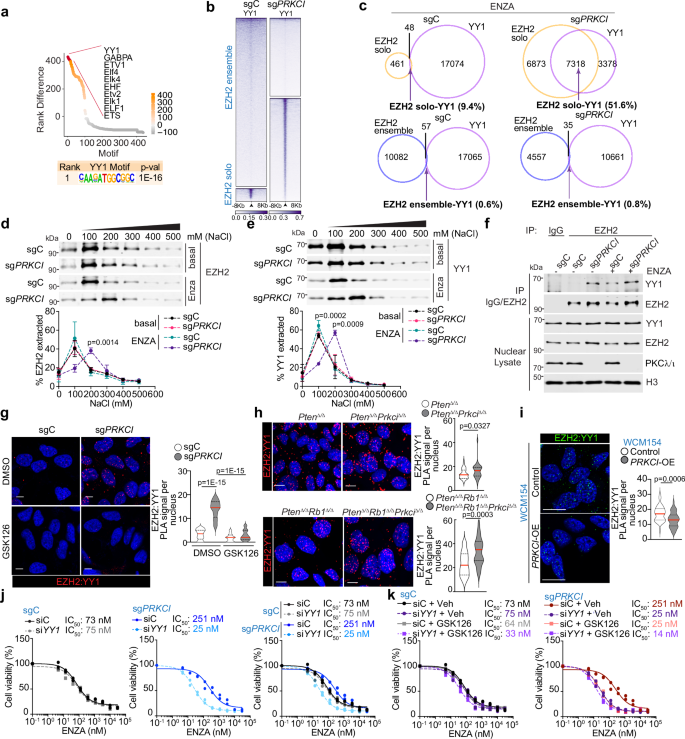
a Enrichment of differential transcription factor motifs between EZH2 solo peaks (all genomic regions) in sgPRKCI and sgC LNCaP cells, plotted by ranks generated from their associated p values (n = 3 biological replicates). b Heatmap of CUT&RUN for YY1 ± 8 kb from the centers of EZH2 solo or ensemble peaks in sgPRKCI and sgC LNCaP cells treated with 10 μM ENZA for 72 h (n = 3 biological replicates). c, Venn diagrams showing the overlap of EZH2 solo or ensemble peaks with YY1 in sgPRKCI and sgC LNCaP cells treated as in (b) (n = 3 biological replicates). d, e Immunoblots and quantification of soluble EZH2 (d) or YY1 (e) extracted using sequential salt extraction assay from sgC and sgPRKCI LNCaP cells treated or not with ENZA (n = 3 independent experiments). f Immunoblots of EZH2 immunoprecipitates in LNCaP cells treated or not with 10 μM ENZA for 72 h (n = 2 independent experiments). g PLA of EZH2 and YY1 in sgC and sgPRKCI LNCaP cells treated or not with 10 μM GSK126 and 10 μM ENZA for 72 h and quantification (sgC-DMSO: n = 40, sgPRKCI-DMSO: n = 41, sgC-GSK126: n = 43, sgPRKCI-GSK126: n = 42 cells examined). Scale bars, 10 μm. h, i PLA of EZH2 and YY1 in PtenΔ/Δ and PtenΔ/ΔPrkciΔ/Δ, PtenΔ/ΔRb1Δ/Δ and PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ mouse prostate organoids (h), or PRKCI-overexpressing (PRKCI-OE) and control NEPC PDOs WCM154 (i), with quantification (PtenΔ/Δ: n = 51, PtenΔ/ΔPrkciΔ/Δ: n = 34, PtenΔ/ΔRb1Δ/Δ: n = 30, PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ: n = 26, PRKCI-OE: n = 42, Control WCM154: n = 41 cells examined). Scale bars, 10 μm. j, k Dose-response curves using CFU assay for 14 days to determine IC50 of ENZA for sgC and sgPRKCI LNCaP cells transduced with the indicated siRNAs or treated with 10 μM GSK126. IC50 value is the average of two biological replicates. Data shown as mean ± SD (d, e) and mean ± SEM (g, h, i) of 3 biological replicates. Two-tailed unpaired Student’s t-test (d, e, g, h, i). Source data are provided as a Source Data file.
To biochemically characterize the EZH2:YY1 complex, we performed a NaCl-based sequential salt extraction assay to determine the effect of ENZA treatment in PKCλ/ι-deficient cells in the affinity of this complex for chromatin binding. Whereas EZH2 and YY1 dissociated maximally from the chromatin between 100-200 mM of NaCl concentration in PKCλ/ι-proficient cells, either treated or untreated with ENZA, there was a shift to the 200–300 mM of NaCl concentration only in ENZA-treated PKCλ/ι-deficient cells (Fig. 6d, e), indicating increased binding of the EZH2:YY1 complex to the insoluble chromatin. RBBP6 knock-down did not influence the affinity of this complex for chromatin binding (Supplementary Fig. 6d, e). ENZA-treated EZH2S375/380AA cells showed a stronger affinity for EZH2 and YY1 towards chromatin than wild-type EZH2-expressing cells, mimicking the phenotype of PKCλ/ι-deficient cells (Supplementary Fig. 6f, g). Endogenous immunoprecipitation and PLA experiments demonstrated the increased EZH2:YY1 interaction in ENZA-treated sgPRKCI cells compared to identically treated sgC cells (Fig. 6f, g). This enhanced interaction was also validated in PKCλ/ι-deficient mouse prostate organoids (Fig. 6h) and was consistently reduced with PRKCI overexpression in WCM154 organoids (Fig. 6i). Of note, treatment with Apalutamide or Darolutamide mimicked the effects of ENZA in terms of chromatin binding and assembly of the EZH2:YY1 complex in PKCλ/ι-deficient cells (Supplementary Fig. 6h–j). Interestingly, treatment with GSK126 inhibitor completely abrogated EZH2:YY1 binding (Fig. 6g), demonstrating that EZH2 solo function also depends on its methyltransferase activity. The functional relevance of the heightened EZH2:YY1 complex was determined in experiments in which YY1 was knocked-down using a specific siRNA. Importantly, the downregulation of YY1 rescued the reduced sensitivity of PKCλ/ι-deficient cells to ENZA, similar to what was observed when ENZA-treated sgPRKCI cells were treated with the EZH2 inhibitor GSK126 (Fig. 6j). Interestingly, treatment of YY1-knocked down PKCλ/ι-deficient cells with GSK126 produced no additional benefit (Fig. 6k), demonstrating that both treatments target the same pathway. These results support the critical role of the EZH2:YY1 complex in the resistance to ENZA promoted by PKCλ/ι loss and unveil a previously unanticipated synthetic vulnerability of this complex to ENZA treatment in the context of PKCλ/ι deficiency.
The non-canonical EZH2:YY1 complex induces a transcriptional program to promote protein translation
To investigate the biological processes regulated by the EZH2:YY1 complex, we integrated the genomic binding data of EZH2 solo peaks and RNA-seq profiles from ENZA-treated PKCλ/ι-deficient cells (Supplementary Fig. 7a, b). Genes activated by the EZH2 solo:YY1 complex were predominantly associated with translation initiation, peptide chain elongation, and pathways related to global translation regulation and ribosomal biogenesis (Fig. 7a). This gene signature was also enriched in primary PCa or CRPC compared to benign tissue when applied to multiple human datasets of PCa30,31,32,33,34,35 (Supplementary Fig. 7b, c). Furthermore, gene set enrichment analysis (GSEA) comparing the transcriptomes of ENZA-treated sgPRKCI and control cells also revealed that translation initiation, elongation, and ribosomes were the highest-ranked gene signatures among the C2-Reactome and C5-GO compilations (Fig. 7b). Consistently, pathway analysis also identified oncogenic MYC targets and mTORC1 signaling as top upregulated hallmarks (Supplementary Fig. 7d). mTORC1 is a well-known master regulator of protein synthesis by controlling ribosome biogenesis and mRNA translation36, and MYC has been shown to influence translation initiation and elongation via mTORC137. The upregulated transcripts dependent on the EZH2 solo binding included genes of the translation machinery such as EIF3 and EIF4 factors, ribosomal genes, and MIOS, a subunit of the GATOR2 complex, critical for mTORC1 activation38 (Fig. 7c, d and Supplementary Fig. 7e). Consistently, immunoblot analysis demonstrated increased phosphorylation of S6K and 4EBP1, two bona fide downstream targets of mTORC1 and of EIF3D, a translation initiation factor transcriptionally upregulated in these cells (Fig. 7e). Knock-down of YY1 reduced the upregulation of the translation machinery and the enhanced mTORC1 activation in PKCλ/ι-deficient cells to control levels (Fig. 7d, e), in agreement with the role of YY1 in mediating EZH2 solo function.
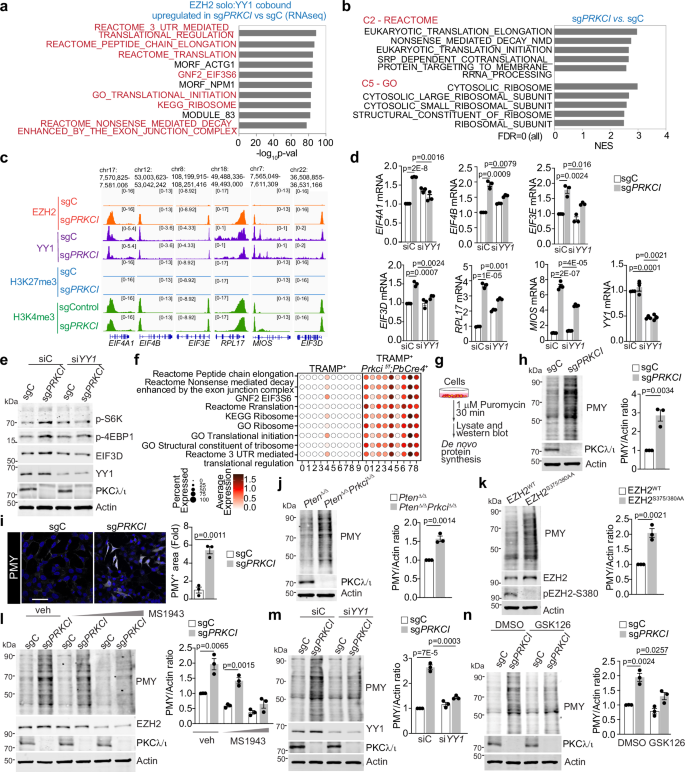
a findGO.pl analysis of upregulated genes that exhibit EZH2 solo cobound with YY1 in sgPRKCI cells treated with 10 μM ENZA for 72 h (n = 3 biological replicates). b GSEA from RNA-seq of sgPRKCI and sgC LNCaP cells treated as in (a) using C2, and C5 gene sets (n = 3 biological replicates). FDR, false discovery rate. c EZH2, YY1, H3K4me3, and H3K27me3 binding (depth normalized) at the indicated loci in sgPRKCI and sgC LNCaP cells treated as in (a) (n = 3 biological replicates). d qPCR from sgPRKCI and sgC LNCaP cells, transduced with siRNAs, and treated as in (a) (n = 3 biological replicates). e Immunoblots in sgPRKCI and sgC LNCaP cells, transduced with siRNAs, and treated as in (a) (n = 2 independent experiments). f Dot plot pathway enrichment map showing the pathway enriched in each cluster of TRAMP+ (n = 1 tumor sample from 1 mouse) and TRAMP+Prkcif/fPbCre+ mice (n = 3 tumor samples from 1 mouse). g Puromycylation assay. Cells were stimulated with 1 μM Puromycin for 30 min, and puromycin-incorporated peptides were detected by immunoblotting. h Puromycylation assay in sgPRKCI and sgC LNCaP cells, treated as in (a), and quantification (n = 3 independent experiments). i Puromycin staining in sgPRKCI and sgC LNCaP cells, treated as in (a) (n = 3 biological replicates). Scale bars, 10 μm. j, k Puromycylation assay in PtenΔ/Δ and PtenΔ/ ΔPrkciΔ/Δ prostate organoids mouse (j) or EZH2WT and EZH2S375/380AA LNCaP cells (k) treated as in (a), and quantification (n = 3 independent experiments). l–n Puromycylation assay in sgPRKCI and sgC LNCaP cells, treated with 10 μM ENZA for 72 h, and 0,2, and 4 μM MS1943 (l), or transduced with the indicates siRNAs (m), or treated with 10 μM GSK126 (n), and quantification (n = 3 independent experiments). Data shown as mean ± SEM of 3 biological replicates (d, h, i, j, k, l, m, n). Two-tailed unpaired Student’s t-test (d, h, i, j, k, l, m, n). Source data are provided as a Source Data file.
To determine whether PKCλ/ι-deficiency in vivo drives this transcriptional program controlling translation, we carried out a single-cell RNA sequencing (scRNA-seq) analysis of prostate tumors from TRAMP+ and TRAMP+Prkcif/fPbCre+ mice. Through unsupervised clustering with specific markers for each population, we identified nine major cell clusters encompassing basal or luminal populations characterized by distinct gene expression patterns, either adenocarcinoma or NEPC-like (Supplementary Fig. 7f–l). Notably, all the epithelial clusters in TRAMP+Prkcif/fPbCre+ mice exhibited enrichment in pathways related to translation and ribosome biogenesis, especially in the basal populations (Fig. 7f and Supplementary Fig. 7m).
To determine whether these transcriptional alterations resulted in changes in protein synthesis rates, we used a puromycin incorporation assay in nascent peptides (Fig. 7g). Both sgPRKCI cells and PtenΔ/ΔPrkciΔ/Δ prostate organoids had higher protein synthesis rates than their respective controls (Fig. 7h–j). Of note, EZH2S375/380AA cells also displayed an increased rate of protein synthesis (Fig. 7k), consistent with the role of EZH2 phosphorylation on the EZH2 solo function. Importantly, EZH2 degradation by MS1943 or YY1 down-regulation rescued the enhanced protein synthesis observed in PKCλ/ι-deficient cells (Fig. 7l, m), further supporting the key role of the EZH2:YY1 complex in promoting protein synthesis in PKCλ/ι-deficient cells. Likewise, treatment with GSK126 also blocked protein synthesis (Fig. 7n), in keeping with the requirement of EZH2 catalytic activity for its non-canonical role.
Increased protein translation accounts for ENZA resistance in PKCλ/ι-deficient cells
We next investigated whether pharmacological inhibition of translation could sensitize PKCλ/ι-deficient cells to ENZA treatment. To that end, we tested tomivosertib (also named eFT50839), an MNK1 inhibitor that blocks translation by reducing eIF4E phosphorylation and activity (Fig. 8a). Interestingly, eFT508 in combination with ENZA had a dramatic synergistic inhibitory effect on the proliferation of PKCλ/ι-deficient cells, whereas eFT508 either alone or with ENZA had barely any effect on control cells (Fig. 8b and Supplementary Fig. 8a). Similarly, translation inhibition by homoharringtonine40 (HTT), an FDA approved drug for refractory CML, or by the mTORC1 inhibitor INK12841, exhibited the same effect as eFT508 in overcoming ENZA resistance in PKCλ/ι-deficient cells (Fig. 8a and Supplementary Fig. 8b, c). Experiments in PKCλ/ι-deficient mouse organoids or human organoids with reduced levels of PKCλ/ι gave similar results regarding the response to the combined treatment of eFT508 and ENZA (Fig. 8c, and Supplementary Fig. 8d–f). In contrast, PRKCI overexpression counteracted the inhibitory effect of this combined treatment on the proliferation of human organoids with reduced PKCλ/ι levels (Fig. 8d). Importantly, the combination of eFT508 and ENZA demonstrated a robust synergistic inhibitory effect on tumor growth in PKCλ/ι-deficient prostate organoids (Ptenf/fTrp53f/fRb1f/fPbCre+) in a xenograft mouse model, without affecting PKCλ/ι-proficient control organoids (Fig. 8e). Overall, these results indicate that the enhanced translation in PKCλ/ι-deficient cells creates a vulnerability that can be exploited therapeutically to increase sensitivity to ENZA.
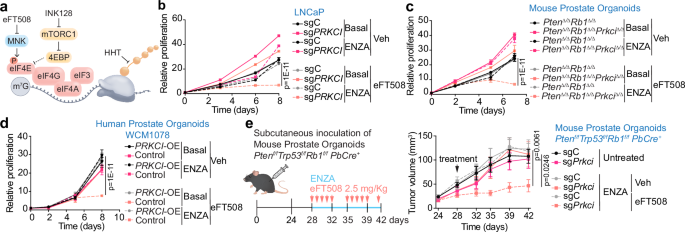
a Mechanisms of action of eFT508, INK128 and HHT inhibitors in translation. b Growth curves of sgC and sgPRKCI LNCaP cells treated with 10 μM ENZA and 10 μM eFT508 alone or combined. Representative experiment from two biological replicates. c Growth curves of PtenΔ/ΔRb1Δ/Δ and PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ mouse organoids treated as in (b). Representative experiment from two biological replicates. d Growth curves of PRKCI-overexpressing (PRKCI-OE) WCM1078 human prostate organoids treated with 10 μM ENZA and 7.5 μM eFT508 alone or combined. Representative experiment from two biological replicates. e Subcutaneous inoculation of mouse prostate Ptenf/fTrp53f/fRb1 f/fPbCre+ organoids in mice treated with ENZA (50 mg/Kg) alone or in combination with eFT508 (2.5 mg/Kg) for 14 days, and tumor volume quantification (sgC-veh: n = 9, sgC-ENZA: n = 9, sgC-ENZA+efT508: n = 8, sgPrkci-veh: n = 8, sgPrkci-ENZA: n = 7, sgPrkci-ENZA+eFT508: n = 7 mice examined). Data shown as mean ± SD of technical triplicates (b, c, d), mean ± SEM (e). Two-way ANOVA (b, c, d, e). Source data are provided as Source Data file.
PKCλ/ι loss increases selective translation to promote a TGFβ resistant program
To better understand the molecular mechanisms whereby PKCλ/ι-mediated translation impacts lineage plasticity and ENZA resistance, we conducted polysome profiling (Fig. 9a). ENZA-treated PKCλ/ι-deficient cells exhibited an increase in the content of 80S monosomes and polysomes (Fig. 9b), indicating an enhanced association of ribosomal subunits during the initiation and elongation steps of protein synthesis. Next, we performed translation efficiency analysis by comparing the levels of ribosome-associated mRNA (polysome RNA-seq) to the total mRNA for each gene. This analysis led to the categorization of genes into three groups: (1) “translation only,” indicating genes with altered polysome RNA-seq but no changes in total RNA; (2) “transcription only,” referring to genes with changes in total RNA but not in polysome RNA-seq; and (3) “homodirectional,” representing genes with alterations in both mRNA and polysome RNA-seq (Supplementary Fig. 9a). Importantly, pathway analysis of the “translation only” group, revealed enrichment in epithelial-mesenchymal transition (EMT), extracellular matrix, and development and neuronal-related pathways (Fig. 9c–e and Supplementary Fig. 9b, c). To validate this analysis, we examined the mRNA and protein levels of the top genes with higher translation efficiency in sgPRKCI. Whereas these genes either did not change or were reduced at the mRNA level (Supplementary Fig. 9d), they were upregulated at the protein level in a YY1- and EIF3D-dependent manner (Fig. 9f, and Supplementary Fig. 9e). These data further support our conclusions that YY1 mediates the increase in translation in PKCλ/ι-deficient cells. EIF3D is a translation initiation factor transcriptionally upregulated by EZH2 solo-YY1 in PKCλ/ι-deficient cells (Fig. 7c) and was previously recognized as a critical factor in the regulation of selective translation42,43. The fact that the knock-down of EIF3D reverted the enhanced translation of EMT and NE targets (such as NCAM1, FSCN1, and TGFB1/2) but had no effect on other targets (such as FOXA1) that do not belong to the “translation only” category regulated in PKCλ/ι-deficient cells (Supplementary Fig. 9e), support that the “selective translation” observed upon loss of PKCλ/ι is due to the regulation of translation initiation through EIF3. Ingenuity Pathway Analysis (IPA) of the genes with differential translation efficiency in PKCλ/ι-deficient cells revealed transforming growth factor β (TGFβ) as the top-ranked predicted upstream regulator (Fig. 9g). Therefore, we next investigated whether pharmacological inhibition of the TGFβ signaling could sensitize PKCλ/ι-deficient cells to ENZA treatment. Remarkably, treatment with TGFβR1-specific inhibitors (galunisertib or A 83-01) overcame the enhanced ENZA resistance in sgPRKCI cells (Fig. 9h–j and Supplementary Fig. 9f), demonstrating the relevant role of TGFβ signaling in the acquisition of ENZA resistance in PKCλ/ι-deficient PCa cells. In summary, our results demonstrate that the loss of PKCλ/ι has a dual effect: first, un-phosphorylated EZH2 exhibits a reduction of its canonical function, promoting the emergence of NEPC features, and second, unphosphorylated EZH2 interacts with YY1, leading to the upregulation of the mRNA translation machinery inducing EMT through TGFβ signaling, ultimately contributing to ENZA resistance (Fig. 9k).
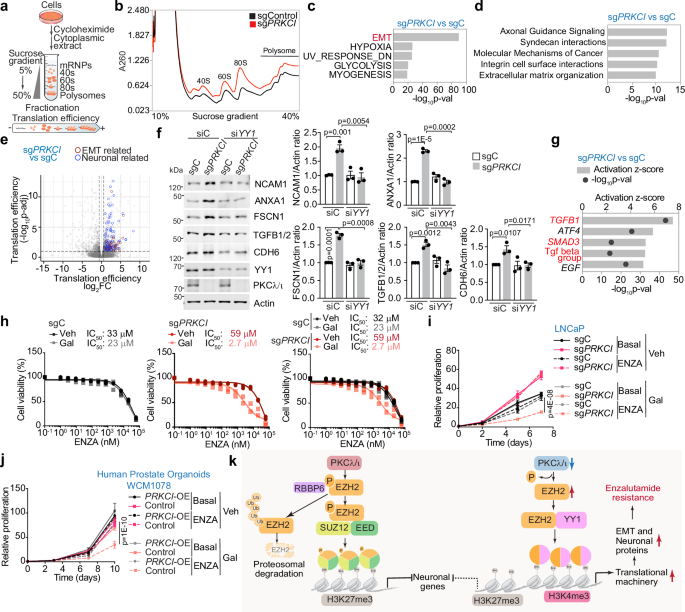
a Schematic of monosome and polysome isolation by sucrose gradient fractionation. b Polysome profiles of sgPRKCI and sgC LNCaP cells treated with 10 μM of ENZA for 72 h (n = 3 biological replicates). c Top 5 Hallmark pathways enriched in translationally efficient mRNAs upregulated in LNCaP sgPRKCI, by HOMER software (n = 3 biological replicates). d Ingenuity Pathway Analysis for translationally efficient mRNAs of LNCaP sgPRKCI vs sgC determined by Xtail (n = 3 biological replicates). e Volcano plot of translationally efficient mRNAs of LNCaP sgPRKCI vs sgC determined by Xtail (n = 3 biological replicates) (Blue= neuronal genes, red= EMT-related genes). f Immunoblots in sgPRKCI and sgC LNCaP cells transduced with the indicated siRNAs, treated as in (a) and quantification (n = 3 independent experiments). g Upstream regulator analysis of translationally efficient genes enriched mRNAs in sgPRKCI, treated with ENZA for 72 h (n = 3 biological replicates). h Dose-response curves to determine the IC50 of ENZA either treated with vehicle or 20 μM galunisertib (Gal) in sgC and sgPRKCI LNCaP cells for 6 days. IC50 value is the average of two biological replicates. i Growth curves of sgC and sgPRKCI LNCaP cells treated with 10 μM of ENZA and 20 μM of galunisertib alone or combined. Representative experiment of two biological replicates. j Growth curves of PRKCI-overexpressing (PRKCI-OE) WCM1078 organoids treated as in (i). Representative experiment of three biological replicates. k PKCλ/ι’s dual role in EZH2 regulation. First, by controlling its stability, mediating its interaction with RBBP6. Second, by facilitating the transition of EZH2 from a Polycomb repressor to a transcriptional coactivator of YY1. This transition mediates resistance to enzalutamide induced by the loss of PKCλ/ι. Data shown as mean ± SEM of 3 biological replicates (f), mean ± SD of technical triplicates (i), mean ± SD of 3 biological replicates (j). Two-way ANOVA (i, j). Two-tailed unpaired Student’s t-test (f). Source data are provided as a Source Data file.
Discussion
Increasing evidence demonstrates that under therapy pressure, tumors undergo a lineage switch driven by epigenetic and metabolic changes, resulting in treatment resistance44,45. However, the detailed signaling mechanisms controlling this process are still largely unknown. Here, we show that PKCλ/ι directly reduces EZH2 levels and function through phosphorylation at S375 and S380, which we demonstrate is a required step for binding to the E3-ubiquitin ligase RBBP6 for its induced ubiquitination and subsequent degradation by the proteasome. Therefore, the loss of PKCλ/ι, a common feature in PCa cells undergoing NE differentiation, results in the accumulation of an unphosphorylated form of EZH2, which we show here is critical for acquiring resistance to ENZA during the adenocarcinoma-to-neuroendocrine lineage transition.
Previous studies in other systems different from the prostate suggested that in response to phosphorylation by several kinases, EZH2 levels can be modulated by the ubiquitin/proteasome system through its interaction with the E3 ligases Smurf2, β-TrCP, FBW7, or Praja146. However, in PCa, the only other study demonstrating the regulation of EZH2 at the protein level implicated the SKP2-induced stabilization of EZH2 through its TRAF6-mediated K63 ubiquitination, independent of the proteasome and likely mediated by the lysosome47. Notably, in addition to the RBBP6-driven mechanism triggered by the S375/S380 phosphorylation by PKCλ/ι reported here, only three other studies have identified EZH2 phosphorylation in PCa, which involved residues S21, T350, or T31121,48,49. Still, none of them regulated EZH2 protein levels, although they reportedly are linked to changes in PRC2 function. Thus, phosphorylation at S21 by AKT or at T350 by CDK1/2 has been shown to switch EZH2 from its repressor role to become a coactivator of AR, promoting gene expression independent of the H3K27me3 mark and reprogramming AR transcriptional activity to induce lineage plasticity21,22. In keeping with the potential clinical relevance of these observations, increased phosphorylation at S21 and T350 has been shown in NEPC samples21,22. In addition, phosphorylation at T311 by AMPK serves to dissociate EZH2 from the PRC2 complex and promote its retention in the cytosol, which facilitates a reversed lineage switch from neuroendocrine back to an AR-luminal stage49.
Our own data establish that PKCλ/ι-mediated phosphorylation of EZH2 at S375/S380 not only maintains the homeostatic levels of EZH2 but also its chromatin repressive function as part of the PRC2 complex. In support of the physiological relevance of this mechanism, we found that EZH2 S380 phosphorylation is reduced in PKCλ/ι-deficient cells in neuroendocrine tumors. Interestingly, our data unveils a previously unappreciated role of PKCλ/ι deficiency in PCa cells on the switch of EZH2 from the PRC2 canonical chromatin-repressive (“ensemble”) complex to a “solo” complex devoid of EED and SUZ12. Such a switch derepresses the transcriptional activation of neuronal and developmental programs while promoting the interaction of EZH2 with YY1 to trigger the transcriptional activation of key regulators of the translation initiation machinery and ribosomal biogenesis. An important upregulated YY1 target includes MIOS, an integral component of the GATOR2 complex, which induces mTORC1 activation, an essential step for translation initiation38. These results should be considered in the context of our previously published data demonstrating the activation of mTORC1 in PKCλ/ι-deficient PCa cells as shown in cell cultures, in vivo models, and human samples16. Those results established that PKCλ/ι phosphorylation of LAMTOR2, a subunit of the Ragulator complex that docks mTORC1 to the lysosomal membrane, was a mechanism for the basal repression of mTORC1 activity16. Therefore, the loss of PKCλ/ι in PCa cells resulted in a heightened mTORC1 activation that drove an ATF4-dependent gene transcription program, resulting in the upregulation of the serine metabolism and increased production of SAM16. Therefore, PKCλ/ι emerges as a central hub in PCa ENZA resistance and lineage plasticity by upregulating translation initiation through the convergence of mTORC1 activity via the direct phosphorylation of LAMTOR2 and the transcriptional upregulation of MIOS by a non-canonical EZH2-driven mechanism. The upregulation of SAM by the ATF4 arm of this pathway likely impinges the methylation of the activating histone marks while producing the substrate for DNA methylation, which we showed previously decisively contributes to the acquisition of the NE phenotype and the subsequent ENZA resistance16.
Therefore, our data advance our understanding of the regulation of mTORC1 in PKCλ/ι-deficient cells by identifying EZH2 phosphorylation as a mechanism to control not only transcription but also translation. Increasing evidence points to the importance of the balance between transcription and translation and the underappreciated role of a dysregulated translational activity in cancer50,51. Of special relevance to our study, previous papers reported the significance of translation in PCa. Thus, the loss of Pten in PCa tumors activates an mTOR-dependent translational program of pro-metastasis mRNAs targetable by mTOR inhibitors41. Also, the aberrant expression and phosphorylation of eIF4E, a key member of the translation initiation complex, was augmented in CRPC, which correlated with poor prognosis52. More recently, an mTOR/eIF4E pathway has been reported to remodel the tumor microenvironment of PCa by rewiring the extracellular matrix (ECM) translatome53. Our findings align with these reports and reveal that PKCλ/ι-deficient cells build up the translational machinery by a non-canonical EZH2-dependent transcriptional mechanism. This enhanced translation capacity results in the increased expression of EMT, ECM, and neuronal proteins regulated by higher translation efficiency in the absence of transcriptional changes and identified TGFβ as their most activated upstream regulator. This is reminiscent of prior reports suggesting the activation of the TGFβ pathway as a major mediator of ENZA resistance54. Our results demonstrate that inhibiting either translation or TGFβ signaling enhances ENZA sensitivity in PKCλ/ι-deficient PCa cells.
These findings are highly relevant for designing future personalized therapeutic approaches and to inform the potential response to EZH2 inhibitors currently evaluated in clinical trials. Our data provide a molecular rationale and identify a synthetic therapeutic vulnerability unique to PKCλ/ι-deficient tumors. These results support an appropriate patient selection based on PKCλ/ι levels that should guide the future evaluation of the efficacy of EZH2 inhibitors in PCa. This is paramount since inhibiting EZH2 will also promote the expression of a set of NE genes basally repressed by the remaining canonical PRC2 complex, which might contribute to therapy resistance. However, the selective blockade of the solo EZH2 arm in PKCλ/ι-deficient tumors, by inhibiting protein translation, not only enhances cell growth inhibition by ENZA but also simultaneously prevents the translation of NE and EMT genes. Therefore, our results highlight that targeting key downstream components of the EZH2 solo program by either blocking TGFβ signaling or inhibiting protein translation will be a more effective therapeutic strategy in restoring ENZA sensitivity than EZH2 catalytic inhibitors.
Methods
Our research complies with all relevant ethical regulations and guidelines. Animal handling and experimental procedures were approved by the Institutional Animal Care and Use Committee at SBP Medical Discovery Institute, and by the Weill Cornell Medicine Institutional Animal Care and Use Committee.
Human samples and ethics approval
Male patients were enrolled in an Institutional Review Board (IRB)-approved protocol with informed consent from Weill Cornell Medicine to build the tissue microarray used in this study. WCM154 PDX slides were provided by Dr. Himisha Beltran.
Mice
Ptenf/fPbCre+, Ptenf/fPrkcif/fPbCre+, Ptenf/fRb1f/fMYCN+PbCre+ mice were previously generated16,25. TRAMP+ (cat No. 003135) and C57BL/6 J (cat No. 000664) mice were purchased from The Jackson Laboratory. TRAMP+ mice were used to generate TRAMP+ Prkcif/f PbCre+, mice. All these mouse strains were generated in a C57BL/6 background. All mice were born and maintained under pathogen-free conditions. Mice were maintained in 24 °C/50% humidity housing subjected to a 14 h light/10 h dark cycle. All genotyping was done by PCR. Age-matched male animals were allocated from each genotype into experimental groups. The endpoint permitted by the ethics committee was 20% of body weight loss. We ensured that each time mice were sacrificed the maximal body weight loss did not exceed this limit. For tissue evaluation n = 3 30-week-old male Ptenf/fPbCre+, Ptenf/fPrkcif/fPbCre+ and TRAMP+ were euthanized following the institutional guidelines. Tissue slides from 14-week-old or 26-week-old castrated Ptenf/fRb1f/fMYCN+PbCre+ were provided by Dr. David Rickman.
Xenograft experiments
For subcutaneous tumor inoculation, Ptenf/fTrp53f/fRb1f/fPbCre+ sgC or sgPrkci organoids were dissociated to a single cell suspension using TrypLE (Thermo Fisher Scientific, cat No. 12604013). 8-week-old C57BL/6J male mice were injected subcutaneously with 1.5 × 106 Ptenf/fTrp53f/fRb1f/fPbCre+ sgC or sgPrkci cells suspended in 1:1 solution of PBS and Matrigel (Corning, #356231). Four weeks after tumor inoculation, tumor size was measured with a caliper and mice were randomized into 3 treatment groups: Untreated, ENZA and ENZA + eFT508 (sgC-veh: n = 9, sgC-ENZA: n = 9, sgC-ENZA+efT508: n = 8, sgPrkci-veh: n = 8, sgPrkci-ENZA: n = 7, sgPrkci-ENZA+eFT508: n = 7). Untreated mice were fed with a regular chow diet (PicoLab Rodent Diet, #5053). Mice receiving ENZA were fed ad libitum with rodent chow supplemented with 50 mg/Kg of ENZA diet (Inotiv). eFT508 (Selleckchem, #S8275) or its vehicle was administered via oral gavage once a day on a Monday through Friday schedule, at a final concentration of 2.5 mg/Kg. Tumor size was assessed biweekly until the experiment concluded. After 2 weeks of drug treatment mice were euthanized following the institutional guidelines. The maximal permitted tumor size of 20 mm in any one dimension was not exceeded in any of our studies.
Cell lines and culture conditions
Cell lines
LNCaP (sex: male; ATCC Cat# CRL-1740, RRID: CVCL_1379), C4-2B (sex: male; ATCC Cat# CRL-3315, RRID: RRID: CVCL_4784), and HEK293T (sex: female; ATCC Cat# CRL-3216, RRID: CVCL_0063) were purchased from ATCC. sgC and sgPRKCI HEK293 cells were previously generated55. Human NEPC organoids WCM1078, WCM1262, and WCM154 were obtained from the Englander Institute for Precision Medicine (EIPM) at Weill Cornell Medicine. Human adenocarcinoma MSKPCa3 organoids26 were obtained from the Memorial Sloan Kettering Cancer Center (MSKCC). Mouse prostate PtenΔ/Δ, PtenΔ/ΔPrkciΔ/Δ, PtenΔ/ΔRb1Δ/Δ, and PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ organoids (sex: male) were generated for this study as described below. Mouse prostate Ptenf/fTrp53f/fRb1f/fPbCre+ organoids (sex: male) were a generous gift from Dr. David W. Goodrich. All cells were negative for mycoplasma, assessed by PCR.
Adenoviral transduction of mouse organoids
Mouse prostate PtenΔ/Δ, PtenΔ/ΔPrkciΔ/Δ, PtenΔ/ΔRb1Δ/Δ, and PtenΔ/ΔRb1Δ/ΔPrkciΔ/Δ organoids were prepared as follows: Normal prostate tissue was dissected from Ptenf/f, Ptenf/fPrkcif/f, Ptenf/fRb1f/f, and Ptenf/fRb1f/fPrkcif/f 15-week-old mice. To prepare prostate cell suspension, prostate tissue was minced in small pieces using a pair of scissors. The prostate pieces were digested with digestion buffer (5 mg/ml Collagenase type II (Thermo Fisher Scientific, cat No. 17101-015), 0.1 mg/ml DNAse (Sigma-Aldrich, cat No. 10104159001), 10 µM Y27632 (Tocris, cat No. 1254) in Advanced DMEM-F12 (Thermo Fisher Scientific, cat No. 12634010) for 60 min at 37 °C with agitation. The prostate cell suspension was washed with 50 ml of Advanced DMEM-F12 completed with 1X HEPES (GIBCO, cat No. 15630080), 1X Glutamax (Thermo Fisher Scientific, cat No. 35050061), 10 µM Y27632, filtered through a 40 μm mesh cell strainer and centrifuged 250 × g for 5 min. Cre-excision of floxed genes was induced in vitro using Adeno-Cre virus (Vector Biosystems, cat No. 1045). Knock-out efficiency was validated by immunoblotting.
Prkci knock-out of mouse organoids
To knock-out Prkci in Ptenf/fTrp53f/fRb1f/fPbCre+ organoids, a single-guide RNA sequence targeting Prkci exon 2 was purchased from Synthego (Supplementary Table 1) and transduced with recombinant Streptococcus pyogenes Cas9 protein (Thermo Fisher Scientific, cat No. A36498), using the Neon Transfection System 1 (Invitrogen) following the manufacturer’s protocol and single clones were expanded and screened by immunoblotting.
Cell culture experiments
LNCaP and C4-2B cells were cultured in Rosewell Park Memorial Institute 1640 (RPMI 1640) (CORNING, cat No. 15-040-CV). HEK293T and sgC and sgPRKCI HEK293 cells were cultured in Dulbecco’s Modified Eagles Medium (DMEM) (CORNING, cat No. 15-013-CV). All base media were supplemented with 10% fetal bovine serum (FBS) (Avantor, cat No. 1300-500), 2 mM glutamine (CORNING, cat No. 25-005-CI), and 100 U/ml penicillin and 100 μg/ml streptomycin (VWR, cat No. 30-002-CI), in an atmosphere of 95% air and 5% CO2. Mouse organoids were cultured in 70% growth factor reduced Matrigel (CORNING, cat No. 356231) in complete organoid media [Advanced DMEM-F12 supplemented with 1X HEPES, 1X Glutamax, 100 U/ml penicillin and 100 μg/ml streptomycin, 10% R-spondin (CM from EIPM), 5% Noggin (CM from EIPM), 1X B27 (Thermo Fisher Scientific, cat No.17504001), 1X N2 (Thermo Fisher Scientific, cat No. 17502048), 50 ng/ml hEGF (Peprotech, cat No. AF-100-15), 200 nM A 83-01 (Sigma-Aldrich, cat No. SML078), 10 µM Y27632, 1 nM 5α-Dihydrotestosterone (DHT) (Sigma-Aldrich, cat No. D073)]. Human organoids were cultured in 70% growth factor reduced Matrigel (CORNING, cat No. 356231) in complete organoid media: Advanced DMEM/F12 (1X) supplemented with 1X HEPES, 1X GlutaMAX, 100 U/ml penicillin and 100 μg/ml streptomycin, 10% R-spondin (Englander Institute of Precision Medicine), 5% Noggin (Englander Institute of Precision Medicine), 1X B27 supplement (ThermoFisher Scientific, cat No. 17504001), 10 mM Nicotinamide (Sigma-Aldrich, cat No. N0636), 1.25 mM N-Acetyl-L-cysteine (Sigma-Aldrich, cat No. A9165), 100 μg/ml Primocin (InvivoGen, cat No. ant-pm-1), 1 ng/ml recombinant hFGF-basic (Peprotech, cat No. 100-18B), 20 ng/ml recombinant hFGF-10 (Peprotech, cat No. 100-26), 1 μM Prostaglandin E2 (R&D Systems, cat No. 2296), 10 μM SB202190 (Sigma-Aldrich, cat No. S7067), 50 ng/ml recombinant hEGF (Peprotech, cat No. AF-100-15), 10 μM Y27632 (R&D Systems, cat No. 1254), 500 nM A 83-01 (Tocris, cat No. 2939), 10 ng/ml recombinant human Heregulinβ−1 (Peprotech, cat No. 100-03) and 1 nM 5α-Dihydrotestosterone (DHT) (Sigma-Aldrich, cat No. D-073).
To stably knock-down PRKCI in C4-2B cells, TRC lentiviral shRNA targeting human PRKCI (Supplementary Table 1) were co-transfected with psPAX2 and pMD2.G packaging plasmids (Supplementary Table 1) into actively growing HEK293T cells using Lipofectamine 2000 (Invitrogen, cat No. 11668-019). Virus-containing supernatants were collected 24, 48, and 72 h after transfection, filtered to eliminate cells, and supplemented with 8 μg/ml polybrene (Santa Cruz Biotechnology, cat No. sc-134220A). Cells were analyzed after 2 μg/ml puromycin (Sigma-Aldrich, cat No. P8833-25MG) selection to confirm knock-down. PRKCI overexpression in WCM154 organoids was performed by infecting WCM154 organoids with lentivirus expressing PRKCI under a tet-inducible promoter (pTRIPZ: Thermo Scientific), followed by selection in 2 μg/ml puromycin 48 h following infection. PRKCI cDNA was cloned intro pTRIPZ as an Age1/Xho1 fragment. To knock-out PRKCI in LNCaP cells single-guide RNA sequences targeting PRKCI exon1 were purchased from Synthego (Supplementary Table 1) and transduced with recombinant Streptococcus pyogenes Cas9 protein (Thermo Fisher Scientific, cat No. A36498), using the Neon Transfection System 1 (Invitrogen) following the manufacturer’s protocol and single clones were expanded and screened by protein immunoblotting. To perform EZH2 editing in LNCaP cells, single-guide RNA sequences targeting the human EZH2 gene were transduced into cells with a Cas9 protein and a single-stranded donor oligonucleotide (Supplementary Table 1) using Neon Electroporation System. Single clones were expanded and screened for EZH2 editing by Sanger sequencing. Knock-down of EZH2, RBBP6, and YY1 was achieved by siRNA transfection with 30 nM of specific siRNA pool using Lipofectamine RNAi MAX transfection reagent (Invitrogen, cat No. 13778030) (Supplementary Table 1). The knock-down efficacy was evaluated 48 h after transfection. Transient overexpression in HEK293T cells was achieved by transfecting 3 μg of different expression plasmids using Lipofectamine 2000. For autophagy inhibition, LNCaP cells were treated with 100 nM bafilomycin A1 or vehicle (DMSO) for 12 h. For proteasome inhibition, LNCaP cell were treated with 50 μM MG132 or vehicle (DMSO) for 12 h. For NEDD8-activating enzyme inhibition, LNCaP cells were treated with 3 μM of MLN4924 or vehicle (DMSO).
Puromycylation assay
Global protein synthesis was analyzed using puromycylation assays as previously described50. Briefly, cells were treated with 1 µM puromycin for 30 min at 37 °C, the whole-cell protein extracts were immediately prepared, and puromycin incorporation into the nascent chain was detected by immunoblotting using a specific anti-puromycin antibody.
Antibodies and reagents
The following antibodies were used in this study: mouse anti-β-actin (#A1978, WB: 1:50,000), mouse anti-FLAG M2 (#F3165, WB: 1:5000), from Sigma-Aldrich; mouse anti-PKCλ/ι, (#610208, WB: 1:1000, IF: 1:200) from BD Biosciences; rabbit anti-HA-Tag (#3724, WB: 1:5000, IP: 1:50), rabbit anti-EZH2 (#5246S, WB: 1:5000, PLA: 1:200, IF: 1:200, IP: 1:50), rabbit anti-YY1 (#46395S, WB: 1:1000, PLA: 1:200), rabbit anti-EED (#85322, WB: 1:5000, PLA: 1:200), rabbit anti-SUZ12 (#3737, WB: 1:1000, PLA: 1:200), rabbit anti-p-S6K (#9205, WB: 1:2000), rabbit anti-p-4EBP1 (#9451, WB: 1:3000), rabbit anti-TGFβ (#3711, WB: 1:500), normal Rabbit IgG (#2729) from Cell Signaling Technology; Phalloidin Alexa Fluor 647 (#A22287, IF: 1:1000), mouse anti-EZH2 (#MA5-18108, WB: 1000, PLA: 1:200), rabbit anti Phospho-EZH2-S380 (custom made-Thermo Fisher Scientific, WB: 1:500, IF: 1:200), goat anti-Mouse IgG1 Alexa Fluor 488 (#A21121, IF: 1:500), donkey anti-Rat IgG Alexa Fluor 488 (#A21208, IF: 1:500), donkey anti-Rabbit IgG Alexa Fluor 568 (#A10042, IF: 1:500) from Thermo Fisher Scientific; mouse anti-UB (#sc-8017, WB: 1:1000), normal mouse IgG (#sc-2025), mouse anti-RPL17 (sc-515904, WB: 1:1000), mouse anti-EIF3E (sc-133251, WB: 1:1000), mouse anti-EIF4B (sc-390912, WB: 1:1000), mouse anti-EIF4A1 (sc-377315, WB: 1:1000), mouse anti-CDH6 (sc-59974, WB: 1:1000) and mouse anti-FOXA1 (sc-101058, WB: 1:1000) from Santa Cruz Biotechnology; rabbit anti-RBBP6 (#a304-975A, WB: 1:500) and mouse anti-EIF3D (#301758, WB: 1:1000) from Bethyl Laboratories; rabbit anti Phospho-EZH2-S21 (#ab84989, WB: 1:1000), rabbit anti-Thiophosphate (#ab92570, WB: 1:5000), rabbit anti-CHGA (ab45179, WB: 1:1000), rabbit anti-Synaptophysin (ab32127, IF: 1:200), rabbit ANXA1 (ab214486, WB: 1:1000), and rabbit anti-H3K27me3 (#ab6002, WB: 1:5000) from Abcam; mouse anti-puromycin (#MABE343, WB: 1:1000) and rabbit anti-NCAM1 (#5032, WB: 1:1000) from Millipore-Chemicon; rabbit anti-MIOS (#20826-1-AP, WB: 1:1000) from Proteintech; goat anti-Rabbit IgG IRDye 800 (#926-32211, WB: 1:5000), goat anti-Mouse IgG1 IRDye 800 (#926-32350, WB: 1:5000), goat anti-Mouse IgG IRDye 800 (#926-32210, WB: 1:5000) from LI-COR Biosciences. Drug treatments, 50 μM MG132 (Selleckchem, cat No.S2619), 100 nM bafilomycin (Baf-A1) (Sigma-Aldrich, cat No. B1793), 50 μg/ml or 100 μg/ml Cycloheximide (Sigma-Aldrich, cat No. C4859-1ML), 1 µM Puromycin (Sigma-Aldrich, cat No. P8833-25MG), 0.1–60 μM Enzalutamide (MDV3100) (Selleckchem, cat No. S1250), 0.1–10 μM GSK126 (Selleckchem, cat No.S7061) 0.1–10 μM EPZ6438 (Selleckchem, cat No. S7128), 0.1–10 μM MS1943 (Selleckchem, cat No.S8918), 0.1–10 μM Tomivosertib (eFT508) (Selleckchem, cat No.S8275), 10 μM Apalutamide (Selleckchem, cat No. S2840), 10 μM Darolutamide (Selleckchem, cat No. S7559), 0.1–20 μM Galunisertib (LY2157299, Cayman Chemical, cat No. 15312), 0.1–10 μM Homoharringtonine (HHT) (MedChemExpress, cat No. HY-14944), 0.1–10 μM A 83-01 (Tocris Bioscience, cat No. 2939), 0.1–10 μM INK128 (Cayman Chemical, cat No. 11811).
Histology, immunohistochemistry, and immunofluorescence
Tissues from indicated mice were isolated, fixed in 10% neutral buffered formalin (Leica, cat No. 3800598) for 12–16 h, dehydrated, and embedded in paraffin. Tissue sections of 5 μm thickness were cut. For the multiplex immunofluorescence, the OPAL™ 4-Color Manual IHC Kit (Akoya Biosciences, cat No. NEL810001KT) was used with fluorophores Opal 520, Opal 570, and TSA Plus Cyanine 5 (Akoya Biosciences, cat No. NEL745001KT), and Spectral DAPI counterstaining. For immunofluorescence on cells, LNCaP were cultured on fibronectin (Sigma, cat No. FC010) pre-treated coverslips and then fixed in 4% paraformaldehyde in PBS (Thermo Fisher Scientific, cat No. J61899.AK) for 15 min at room temperature and then washed with PBS. Antigen retrieval was performed using 10 mM ammonium chloride for 10 min, then washed three times in PBS. Cells were permeabilized with 0.3% Triton in PBS for 10 min and then washed three times in PBS. Cells were blocked 1% BSA in PBS 1 h, and then incubated with the diluted primary antibody in 1% BSA in PBS overnight at 4 °C, followed by incubation with Alexa-conjugated secondary antibodies for 2 h and with DAPI for 15 min. Proximity Ligation Assay (PLA) was performed using Duolink® In Situ Detection Reagents Red (Sigma-Aldrich, cat No. DUO92008), Duolink® In Situ PLA® Probe Anti-Rabbit PLUS (Sigma-Aldrich, cat No. DUO92002), and Duolink® In Situ PLA® Probe Anti-Mouse MINUS (Sigma-Aldrich, cat No. DUO92004), following the manufacturer’s protocols.
Tissue microarray and histological analyses
Scanned TMA slides were examined and those cores with either no tumor represented or with artefacts (tissue broken, for example) were excluded. PKCλ/ι and EZH2 expressions in human samples were evaluated by intensity of staining and percentage of stained cancer cells: for PKCλ/ι intensity was given scores 0–3 (0 = no, 1 = weak 2 = moderate, 3 = intense), and the percentage of positive cells was given scores 0–4 (0 = 0%, 1 = 1–25%, 2 = 26–50%, 3 = 51–75%, 4 = 76–100%). The two scores were multiplied to obtain the result of 0–12. Expressions were considered positive when scores were more than 6 and negative when scores were 6 or less. For EZH2, samples were considered positive when nuclear signal was detected, otherwise was consider negative.
Immunoblotting and immunoprecipitation assay
Cells for protein analysis were lysed in RIPA buffer (20 mM Tris-HCl, 37 mM NaCl, 2 mM EDTA, 1% Triton-X, 10% glycerol, 0.1% SDS, and 0.5% sodium deoxycholate) with phosphatase and protease inhibitors or 1% SDS buffer (1% SDS, 10 mM Tris-HCl, 1 mM EDTA) with sonication. For immunoprecipitation in total fraction, cells were lysed in IP lysis buffer (150 mM NaCl, 20 mM Tris-HCl, 0.5% Igepal, 1 mM EDTA, 1 mM EGTA) with phosphatase and protease inhibitors. Cell lysates were incubated with the corresponding antibodies overnight and immunoprecipitated with 20 μl of 50% slurry of protein G-Sepharose 4B (Invitrogen, cat No. 101242). Immunoprecipitates were washed several times with wash buffer (lysis buffer with 150 mM NaCl). For immunoprecipitation from the nuclear fraction, cells were incubated with EB0 buffer (50 mM Tris-HCl at pH 7.5, 1 mM MgCl2, 0.1% Igepal, 1 mM EDTA, with phosphatase and protease inhibitors) for 10 min on ice, centrifuged at 2500 × g. Cytoplasmic fraction was discarded, and nuclei were resuspended in EB300 buffer (50 mM Tris-HCl at pH 7.9, 1 mM MgCl2, 300 mM NaCl, 1 % Igepal, 0.2 mM PMSF, 1 mM EDTA with phosphatase and protease inhibitors), incubated for 30 min on ice, and centrifuged at 20,000 × g. Protein concentration of the lysates was determined by using Protein Assay Kit (Bio-Rad, cat No. 5000115, 5000113, 5000114). Cell extracts and immunoprecipitated proteins were denatured, subjected to SDS-PAGE, transferred to PVDF membranes (Millipore, cat No. IPFL00010). After blocking with Intercept Protein-Free Blocking buffer (LI-COR, cat No. 927-80001), the membranes were incubated with the specific antibodies overnight at 4 °C. After 2 h incubation with the appropriate horseradish peroxidase-conjugated antibodies, the immunocomplexes were detected by chemiluminescence (Thermo Scientific) or Near-infrared fluorescence (LI-COR). To detect endogenous EZH2 ubiquitination, cells were lysed with Ubiquitination lysis buffer (2% SDS, 150 mM NaCl, 10 mM Tris-HCl, pH 8.0, with 2 mM sodium orthovanadate, 50 mM sodium fluoride, and protease inhibitors). Cell lysates were boiled for 10 min to dissociate protein-protein interactions. The samples were diluted with dilution buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% Triton), and an immunoprecipitation assay was performed as described above. For an example of the presentation of full scan blots, see the Source Data file.
Mass spectrometry analysis
HA-EZH2 plasmid was transfected to ~80% confluent sgC and sgPRKCI HEK293 cells. For nuclear protein extraction, cells were incubated with buffer A (20 mM Tris-HCl at pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 mM PMSF) for 10 min on ice, centrifuged at 2500 × g, resuspended in buffer C (20 mM Tris-HCl at pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.5 mM DTT, 0.2 mM PMSF, 0.2 mM EDTA), sonicated, and centrifuged at 20,000 × g for 30 min. HA-EZH2 were subsequently immunoprecipitated with anti-HA antibody from HEK293 sgC (n = 1) and sgPRKCI (n = 1) and eluted using HA-peptide (Thermo Scientific). 5 μg of eluted proteins from each set were used for mass spectrometry analysis. For the in vitro phosphorylation assays, 3 μg of EZH2 was incubated either with (n = 1) or without (n = 1) 1.5 μg of purified PKCλ/ι (Thermo) at 30 °C for 60 min in 100 μL of kinase assay buffer containing 200 μM of ATP, 1 mM dithiothreitol (DTT), 25 mM HEPES (pH 7.4) and 15 mM of MgCl2. Samples were subsequently analyzed using mass spectrometry to identify EZH2 interacting partners or phosphopeptides altered in PKCλ/ι-depleted cells (in-cell phosphorylation). Protein digestion, TiO2-based phosphopeptide enrichment, electrospray ionization-liquid chromatography-tandem mass spectrometry, and MS/MS analysis were performed as described previously56. The samples were digested with trypsin overnight at 37 °C following reduction with 5 mM DTT and alkylation with 14 mM iodoacetamide. The digests were vacuum centrifuged to dryness and desalted by C18 micro-columns prior to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. For phosphopeptide analysis, the peptide samples were reconstituted in 3% trifluoroacetic acid (TFA)/60% acetonitrile and passed through micro-columns packed with TiO2 beads. The TiO2 beads were washed once with 3% TFA/80% acetonitrile, once with 3% TFA/30% acetonitrile, and once with 0.1% TFA/80% acetonitrile. Phosphopeptides were eluted from the beads with 3% ammonia hydrate (pH 10, diluted from a 28% ammonia–water solution) and 1.5% ammonia hydrate/50% acetonitrile. An EASY-nLC 1200 UPLC (Thermo Fisher Scientific) coupled on-line to a Fusion Lumos mass spectrometer (Thermo Fisher Scientific) was used for all LC-MS/MS analyses. Buffer A (0.1% FA in water) and buffer B (0.1% FA in 80% ACN) were used as mobile phases for gradient separation. A 75 µm × 15 cm chromatography column (ReproSil-Pur C18-AQ, 3 µm, Dr. Maisch GmbH, German) was packed in-house for peptide separation. Peptides were separated with a gradient of 3–40% buffer B over 50 min, 40–100% B over 10 min at a flow rate of 300 nL/min. The Fusion Lumos mass spectrometer was operated in data dependent mode. Full MS scans were acquired in the Orbitrap mass analyzer over a range of 300–1500 m/z with resolution 70,000 at m/z 200. The top 20 most abundant precursors with charge states between 2 and 5 were selected with an isolation window of 1.4 m/z by the quadrupole and fragmented by higher-energy collisional dissociation with normalized collision energy of 35. MS/MS scans were acquired in the Orbitrap mass analyzer with a resolution of 15,000 at m/z 200. The automatic gain control target value was 1e6 for full scans and 5e4 for MS/MS scans, respectively, and the maximum ion injection time was 100 ms for MS scans and 54 ms for MS/MS scans. The raw files were processed using the MaxQuant57 computational proteomics platform version 1.6.17.0 (Max Planck Institute, Munich, Germany) for protein and peptide identification. The fragmentation spectra were used to search the UniProt human protein database (downloaded on 09/21/2017). Oxidation of methionine, protein N-terminal acetylation, and phosphorylation on serine, threonine, and tyrosine were used as variable modifications for database searching. Both peptide and protein identifications were filtered at 1% false discovery rate based on decoy search using a database with the protein sequences reversed.
In vitro kinase-assay and MS/MS phosphopeptide identification
HA-tagged human EZH2 (HA-EZH2) plasmid was transfected to ~80% confluent HEK293T cells in a P100 format. The amount of EZH2 mutants transfected was corrected to equal levels in the expression. Cells were lysed in RIPA buffer 48 h after transfection and HA-EZH2 was immunoprecipitated using anti-HA beads (Thermo Fisher Scientific, cat No. 26181). Immunoprecipitates were washed and incubated at 30 °C for 60 min in kinase-assay buffer containing 25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.5 mM EGTA, 1 mM DTT, and 400 μM ATPγS (Abcam, cat No. ab138911) or 200 μM ATP in the presence of recombinant PKCλ/ι (Thermo Fisher Scientific, cat No. PV3186). Detection of substrate phosphorylation was performed using the ATP analog-based phosphorylation detection used previously55 with minor modifications. Briefly, after the phosphorylation reaction, PNBM (Abcam, cat No. ab138910) and EDTA were added to a final concentration of 2.5 mM and 20 mM, respectively, and incubated for 1 h at room temperature. Immunoblotting detection was performed with anti-thiophosphate ester antibody. To identify phosphorylation sites on EZH2 mediated by PKCλ/ι, we employed both in vitro and in-cell phosphorylation assays coupled with a MS/MS approach. The in vitro phosphorylation assay, in-cell phosphorylation assay, and MS/MS analysis were performed as described above. Phosphorylation sites of EZH2 by PKCλ/ι were identified by filtering hits from the mass spectrometry analyses based on several criteria. These criteria included conservation across different species, exclusion of proline-directed phosphorylation sites, surface accessibility (measured by NetSurfP software, with a threshold above 0.3), and fold changes in intensity ratios. Specifically, for the in vitro phosphorylation assay, the fold change was determined as EZH2 + PKCλ/ι vs EZH2 alone. For the in-cell phosphorylation assay, it was EZH2 in control cells vs EZH2 in PKCλ/ι-depleted cells, with a minimum fold change of 1.25. Hits that met these filtering criteria from both mass spectrometry approaches were compared to identify common phosphorylation sites. These common sites were then selected for subsequent validation.
Drug treatment and cell proliferation assay
sgC and sgPRKCI LNCaP and C4-2B cells were trypsinized into single-cell suspension using 0.25% trypsin supplemented with 2.21 mM EDTA. The trypsin was quenched with two volumes of RPMI 1640 supplemented with 10% FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, and live cells were counted in a hemocytometer using trypan blue exclusion staining. 250 live cells were seeded in each well of a flat-bottomed, 384-well tissue culture-treated microplate in quintuplicate and incubated overnight in a humidified atmosphere of 5% CO2 at 37 °C. To assess their proliferation, organoids were dissociated into single-cell suspension by gentle shaking using TrypLE reagent for 15 min. The cells were washed once in 1X calcium and magnesium-free Dulbecco’s Phosphate buffered saline (DPBS) (CORNING, cat No. 21-031-CV) and resuspended in organoid media. 250 live cells per well were seeded in 70% growth factor reduced Matrigel in organoid media in each well of a flat-bottomed, 384-well tissue culture treated microplate and incubated overnight in a humidified atmosphere of 5% CO2 at 37 °C. After overnight incubation, specific doses of the inhibitors were added to each well either alone or in combination with ENZA and Cell Titer Glo assay (Promega, cat No. G9682, G7570) was performed at specific time points according to the manufacturer’s protocol. The luminescence signal generated from each well was acquired using a microplate reader. The raw luminescence value from each well was normalized to the mean of corresponding wells treated with 0.1% DMSO, and the relative proliferation was measured by plotting the data using GraphPad Prism software.
Assessment of drug synergism and IC50 using CFU assay
sgC and sgPRKCI LNCaP and C4-2B cells were trypsinized, and single-cell suspension was prepared. The live cells were counted using trypan-blue exclusion staining in a hemocytometer and 40 cells were seeded per well of a flat-bottomed 96-well plate. After overnight incubation, a 10-point serial dilution of the compounds was prepared using 1:3 dilution and added to subsequent wells in a final volume of 200 μL of complete media. The cells were incubated for another 2 weeks without changing the media and the colonies were detected after staining with crystal violet (Sigma Aldrich, cat No. V5265). Crystal violet staining was performed by washing the colonies with 0.9% NaCl after removal of the culture media. The colonies were fixed for 20 min in 10% neutral buffered formalin and stained with 0.01% (w/v) crystal violet for 30 min. The stain was removed, and the colonies were washed once with distilled water. Finally, the stain was dissolved by incubating the colonies in 100 μL of 10% acetic acid (v/v) for 30 min with gentle rocking and the optical density for each well was measured at 595 nm in a Bio-Rad plate reader. The dose response curves were determined by non-linear regression method and the IC50 values were calculated using GraphPad Prism. To determine the synergistic effects of two drug combination, the cells were seeded in triplicate and treated with increasing doses of either drug alone or in combination and cultured for 2 weeks without refreshing the media. After staining the colonies with crystal violet, the excess stain was washed once with distilled water. The stain was dissolved in 100 μL of 10% acetic acid and the optical density (O.D) was measured at 595 nm. The synergy score was determined by Bliss synergy model using SynergyFinder 2.2 and the data were represented as percent inhibition relative to the vehicle.
Assessment of drug synergism and IC50 using cell proliferation method
To determine the IC50 by cell proliferation method, 1000 live cells per well were seeded in each well of a flat-bottomed, 384-well tissue culture-treated microplate and incubated overnight in a humidified atmosphere of 5% CO2 at 37 °C. After overnight incubation, a 10 or 12-point serial dilution of ENZA was prepared using 1:3 dilution intervals spanning 60 μM to 0.1 nM and added to subsequent wells either in the presence or absence of specific compounds in a final volume of 80 μL of complete media. The cells were incubated for another 6 days while the media was refreshed with the compounds after 3 days of incubation. The live cells were stained with CellTiter Glo (Promega) according to the manufacturer’s protocol, and the luminescence signal generated from each well was acquired using a microplate reader. The raw luminescence value from each well was normalized to the mean of corresponding wells treated with 0.1% DMSO and the IC50 values were calculated using GraphPad Prism by non-linear regression method.
Sequential salt extraction assay
10 × 106 LNCaP cells were washed twice with 5 ml of ice cold 1X DPBS, resuspended in 1 ml of hypotonic Buffer A (0.3 M sucrose, 60 mM KCl, 60 mM Tris at pH 8.0, 2 mM EDTA and 0.5% NP-40) supplemented with protease inhibitor cocktail and incubated at 4 °C for 10 min with end-to-end rotation. The nuclei were isolated by centrifugation at 6000 × g at 4 °C for 10 min and resuspended in 100 μL of modified RIPA buffer (100 mM Tris at pH 8.0, 2% NP-40 and 0.5% sodium deoxycholate) without NaCl. The nuclei were homogenized by pipetting 15 times and incubated on ice for 5 min. The chromatin pellet was isolated by centrifugation at 10,000 × g for 5 min at 4 °C and the supernatant representing 0 mM fraction was collected. This process was repeated with the nuclei treated sequentially with increasing NaCl concentration and the soluble fractions corresponding to 100, 200, 300, 400, and 500 mM NaCl were collected. To determine the binding profile of the proteins, equivalent volumes from each fraction was analyzed by immunoblot and quantitative densitometry was performed using LI-COR Odyssey imaging system. To analyze the percent protein eluted at each fraction, band intensities from each fraction were compared to the cumulative band intensities from all the fractions.
Gene-expression analyses
Total RNA was extracted by using TRIzol reagent (Thermo Fisher Scientific, cat No. 15596018) and purified by using Quick-RNA Mini Prep Kit (Zymo Research, cat No. R1054) following the manufacturer’s protocols. After quantification using a Nanodrop 1000 spectrophotometer (Thermo Scientific, 1 μg of RNA was reverse transcribed using random primers and MultiScribe Reverse Transcriptase (Thermo Fisher Scientific, cat No. 4311235). Gene expression was analyzed using the CFX96 Real Time PCR Detection System with SYBR Green Master Mix (Bio-Rad, cat No. 1725125). Primer sequences are listed in Supplementary Table 2. The amplification parameters were set at 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s (40 cycles total). Gene expression values for each sample were normalized to the 18 s rRNA.
CUT&RUN assay and sequencing
Cleavage under targets and release using nuclease (CUT&RUN) assay was performed using the CUTANA ChIC/Cut&Run kit v3.5 from Epicypher. Before performing the assay, all the required buffers were prepared according to the manufacturer’s protocol. 10 μL of ConA beads per reaction were washed twice with bead activation buffer and kept on ice. For each sample out of the triplicates, including the positive and negative antibody controls, 500,000 cells were harvested after dissociation using StemPro Accutase, washed twice with wash buffer, and incubated with the Concanavalin A (ConA) beads for 10 min at room temperature. Post incubation, the ConA bound cells were resuspended in antibody buffer and 0.5 μg of specific antibodies including negative IgG and H3K4me3 positive control antibodies, and mixed with K-Metstat internal control panel and incubated overnight on a nutator at 4 °C. The following antibodies were used: CUTANA™ IgG Negative Control Antibody for CUT&RUN and CUT&Tag (#13-0042, 1:50), rabbit anti-H3K4me3 SNAP-Certified™ for CUT&RUN (#13-0041, 1:50), rabbit anti-EZH2 CUTANA™ CUT&RUN (#13-2026, 1:50), rabbit anti-H3K27me3 SNAP-Certified™ for CUT&RUN and CUT&Tag (#13-0055, 1:50) from Epicypher and rabbit anti-YY1 (#46395, 1:50) from Cell Signaling Technology. The cells were washed twice with cell permeabilization buffer containing 0.025% digitonin and incubated with protein A/protein G (pAG) bound micrococcal nuclease (pAG-MNase) for 15 min at room temperature. The cells were washed with cell permeabilization buffer and mixed with 2 mM calcium chloride to induce nuclease-mediated chromatin digestion for 2 h and 30 min at 4 °C. The digestion reaction was terminated by the addition of Stop master mix containing 0.5 μg of E.coli spike-in DNA and the incubation at 37 °C for 10 min. To purify the fragmented DNA, the binding buffer was added to each reaction and collected by centrifugation. The DNA was washed twice with wash buffer, elution was done in 50 μL of 0.1X TE buffer, 5 ng of purified CUT & RUN DNA was used to prepare libraries for sequencing using NEBNext Ultra II DNA Library Prep Kit for Illumina. 5’-phosphorylation and 3’-dA tailing were done using End Prep enzyme, and the samples were incubated at 20 °C for 30 min, followed by 65 °C in a thermal cycler. Each sample of fragmented DNA was adapter ligated and purified using 0.9X volume of SPRIselect beads and dissolved in 15 μL of 0.1X TE buffer. Each adapter ligated DNA sample was PCR enriched using universal PCR primer and specific Index primer and the amplified DNA were cleaned up with 0.9X SPRIselect beads. Finally, the DNA libraries were dissolved in 30 μL of 0.1X TE buffer and the size distribution was checked on an Agilent Bioanalyzer High sensitivity chip and quantified using Invitrogen Qubit Fluorometer. Barcoded libraries were pooled, and single end sequenced (2 × 150 bp, paired end) on the Illumina HiSeq 4000 at AZENTA, LLC.
RNA-seq preparation and sequencing
Total RNA was extracted using the Quick-RNA MiniPrep kit (Zymo Research). Library preparation was performed using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina following the manufacturer’s instructions (New England Biosystems) at AZENTA, LLC. Barcoded libraries were pooled and single-end sequenced (2 × 150 bp, paired end) on the Illumina NovaSeq 6000 at AZENTA, LLC.
Polysome profiling and sequencing
Polysome profiling was performed as described58 with minor modifications. Briefly, sgC and sgPRKCI LNCaP cells were seeded in two 15-cm dishes. At 70% confluence, cells were treated with 100 μg/ml cycloheximide (Sigma, C7692) 15 min before collection. After two washes in ice-cold PBS containing cycloheximide (100 μg/ml), cells were scraped in polysome lysis buffer (30 mM Tris-HCl, pH 7.5, 100 mM NaCl, 30 mM MgCl2, 1% sodium deoxycolate, 1% Triton X-100, 100 μg/ml cycloheximide, 1 mM dithiothreitol (DTT) and 30 U ml−1 RNasin). Cytoplasmic extracts with equal amounts of RNA were loaded on a 10–45% sucrose gradient and centrifuged at 4 °C for 3 h at 190,000 × g in a SW41Ti Beckman rotor using a Beckman Optima L-90K ultracentrifuge. Gradients were read at 260 nm by the BioLogic LP system (BioRad) and acquired by the TriaxTM FlowCell software. The fractions (1.0 ml each) were collected for subsequent RNA extraction to isolate the polysomal RNA fractions. Total RNA was extracted by using TRIzol reagent (Invitrogen) and purified by using Quick-RNA Mini Prep Kit (Zymo Research) following the manufacturer’s protocols. The purified RNA was measured using Nanodrop and the quality was assessed using the Agilent 2100 Bioanalyzer. Library preparation was performed using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina following manufacturer’s instructions (New England Biosystems) at AZENTA, LLC. Barcoded libraries were pooled, and single end sequenced (2 × 150 bp, paired end) on the Illumina NovaSeqX.
10x library preparation and sequencing
To prepare prostate tumor cell suspension, tumors were digested with digestion buffer (5 mg/ml Collagenase type II in Advanced DMEM-F12 containing 10 µM Y27632) for 1 h at 37 °C with agitation, washed with Advanced DMEM-F12 completed with Hepes 1x, Glutamax 1x, 10 µM Y27632 and 5% FBS (ADF 4+), filtered through 70 and 40 μm mesh cell strainers, and resuspended with ADF 4+. This was followed by a 20 min digestion with TryPLE at 37 °C. Cells were washed as before and dead cells were removed by EasySep Dead Cell Removal (Annexin V) kit (STEMCELL technologies). scRNA-seq libraries were generated using the Chromium Single Cell 30 Reagent Kit v2 (10X Genomics). Cells were loaded onto the 10x Chromium Single Cell Platform (10x Genomics) at a concentration of 2000 cells per µL (Single Cell 3’ library and Gel Bead Kit v.2) as described in the manufacturer’s protocol (10x User Guide, Revision B). Generation of gel beads in emulsion (GEMs), barcoding, GEM-RT clean-up, complementary DNA amplification, and library construction were all performed as per the manufacturer’s protocol. Individual sample quality was checked using a Bioanalyzer Tapestation (Agilent). Qubit was used for library quantification before pooling. The final library pool was sequenced on an Illumina NovaSeq6000 instrument using an S1 flow cell.
Bioinformatics analysis of International SU2C/PCF Dream Team Dataset24
Patients were classified into low, moderate, and high categories for PRKCI expression based on fixed sample thresholds—specifically selecting the top and bottom 30 samples for the high and low categories, respectively. To validate and visually represent this classification, we superimposed the thresholds on a normal distribution fit of the entire dataset’s PRKCI expression levels. Detailed statistical analysis revealed that the densities at the low and high thresholds are ~0.037 and 0.032, respectively. These density values, derived from the probability density function of the normal distribution, confirm that the chosen thresholds effectively mark meaningful shifts within the data distribution. These values underscore that the chosen cut-offs capture potential biologically relevant variations in expression levels. Pearson correlation was used for pairwise-comparisons of PRKCI and the represented genes.
CUT&RUN analysis
For CUT&RUN experiments, quality control of raw sequencing reads was performed using FastQC (Babraham Bioinformatics). Low-quality reads were removed using Trimmomatic59 with a sliding window size of 4 bp and a quality threshold of 20. Paired-end Homo Sapiens reads were mapped to UCSC GRCh38 using Bowtie260 version 2.2.4 with parameters –end-to-end –verysensitive –no-mixed –no-discordant -q –phred33 -I 10 -X 700. PCR duplicates introduced during library creation were removed using SAMtools61. Peak calling was performed using MACS2 with a q-value threshold of 0.0001 for EZH2 and H3K4me3, and 0.01 for YY1. For H3K27me3, the broad-peak option was enabled with a broad-cutoff q-value threshold of 0.1. Sequencing reads from IgG precipitates in each condition were utilized as controls during peak calling. To compare binding across different samples, we employed MACS2 bdgdiff, which accounts for the depth of sequencing across conditions. For visualization, bigwig files were generated using the MACS2 callpeak function with the --SPMR command, which normalizes the signal to million reads. Sequencing reads from IgG immune precipitates in each condition were used as a control for peak calling. Peaks were annotated to hg38 genomic features and assessed for the presence of transcription factor motifs using HOMER62. Regions of CUT&RUN overlap were defined using BEDTools63 and Venn diagrams were plotted using VennDiagram R package. Uniquely bound EZH2 solo and ensemble peaks per genotype were intersect with YY1 signal using BEDTools63. PCA plots, enrichment profile plots and heatmaps were generated using deepTools64. findGO.pl function from HOMER was used to do pathway analysis. The integration of CUT&RUN data with RNA-seq was done as follows: a 329-gene signature was generated by integrating RNA-seq data from ENZA-treated, PKCλ/ι-deficient LNCaP cells with CUT&RUN data to identify significantly upregulated genes (adjusted p < 0.05, log2 fold change >0.3) that exhibited EZH2 solo signal cobound with YY1 at their promoters. Subsequently, we analyzed this gene signature in RNA-seq data from human PCa patients using GSEA.
RNA-seq analysis
Raw sequence data (.bcl files) generated were converted into fastq files and de-multiplexed using Illumina bcl2fastq 2.20 software. Quality control of raw sequencing reads was performed using FastQC (Babraham Bioinformatics). Sequencing Fastq files were trimmed using the bbduk program from BBTools (Joint Genome Institute, https://sourceforge.net/projects/bbmap/) to remove low-quality bases at ends, and the resulting reads were mapped to the reference genome (GRCh38) using STAR65 with –quantMode GeneCounts. Reads were sorted and indexed using SAMtools61. Differential Expression analysis for RNA-Seq data was performed using R/Bioconductor package DESeq266. Genes characterized by low mean normalized counts were filtered out by the independent filtering feature embedded in DESeq2. Gene Set Enrichment Analysis (GSEA) was performed using GSEA v4.3.2 software (http://www.broadinstitute.org/gsea/index.jsp) using default parameters with MSigDB h.all.v2023.1.symbols (H), c2.all.v2023.1.symbols (C2) and c5.all.v2023.1.symbols (C5) collections or customized signatures (Supplementary Data 1).
Polysome profiling RNA-seq analysis
The polysomal RNA and total RNA expression for each genetic background were compared to the wild-type. Xtail67 package was used to infer the translational efficiencies and perform differential analysis. Volcano plots were made using the R package EnhancedVolcano. (https://github.com/kevinblighe/EnhancedVolcano).
Single-cell RNA-seq analysis
Raw sequence reads were quality-checked using FastQC software. The Cell Ranger version 6.0 software suite from 10X Genomics (https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest) was used to process, align, and summarize unique molecular identifier (UMI) counts against the mouse mm10 assembly reference genome analysis set, obtained from the University of California Santa Cruz (UCSC). Raw, unfiltered count matrices were imported into R for further processing. Raw UMI count matrices were filtered using the Seurat v 4.0 R package68 to remove: barcodes with very low (less than 200, empty wells) and very high (more than 4000, probably doublets) total UMI counts; matrices for which a high percentage of UMIs originated from mitochondrial features (more than 12%); and matrices for which fewer than 250 genes were expressed. Subsequently, the data were normalized using the SCTransform function, regressing out the following variables: total number of UMIs per cell and percentage of mitochondrial UMIs. Following normalization, the principal components were computed. The top principal components were identified using the ElbowPlot function and used for the UMAP dimensionality reduction. For clustering, we used RunUMAP(), FindNeighbors(), and FindClusters() functions for clustering and the percentage of mitochondrial features was considered to be a source of unwanted variation and regressed out using the Seurat package. Genes specifically expressed in each cluster were identified with the FindAllMarkers() function and the Wilcoxon test labeling the different populations using the genes differentially up-regulated in each population. Epcam-negative cells were not considered, and the cell groups were annotated based on the marker gene analysis and canonical markers from the literature. The scoring for the indicated signatures was performed using the AddModuleScore function in Seurat with default parameters. Gene sets used for signature scoring are listed in Supplementary Data 1.
Statistics and reproducibility
All the statistical tests were justified for every figure. All samples represent biological replicates. Immunoblot experiments were repeated at least two times independently with similar results. Data are presented as the mean ± SEM. Statistical analyses were performed using GraphPad Prism 9 or R software environment (http://www.r-project.org/). Significant differences between groups were determined using a student’s t-test (two-tailed) when the data met the normal distribution tested by D’Agostino test. If the data did not meet this test, a Mann-Whitney U-test was used. Differences between more than 3 groups were determined using one-way ANOVA test (parametric) or Brown-Forsythe or multiple t-test and Welch ANOVA tests (nonparametric) followed by Tukey’s or Bonferroni’s post hoc test. The chi-square test or Fisher’s exact test was used to determine the significance of differences between covariates. Logistic regression analysis was employed to estimate univariate and multivariate odds ratio and 95% confidence interval (CI). Values of p < 0.05 were considered as significantly different. No data were excluded from the analyses. No statistical method was used to predetermine the sample size. Investigators were not blinded to group allocation at the time of data collection and analysis. For each experiment, two or three replicates were used. All attempts at replication generated reproducible results supporting the overall conclusion.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq, Polysome profiling, CUT&RUN and single-cell RNA-seq data generated in this study have been deposited in the public repository GEO (Gene Expression Omnibus) database under accession code GSE266956. Proteomics data generated in this study have been deposited in MassIVE with identifiers MSV000094704, MSV000094705, MSV000094706.The publicly available data used in this study are available in GitHub https://github.com/cBioPortal/datahub/tree/master/public/prad_su2c_201924 and in the GEO database under accession codes GSE18137426, GSE8922330, GSE7076831, GSE10474932, GSE2820433, GSE537734, GSE8060935. The remaining data are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
References
Mateo, J. et al. Accelerating precision medicine in metastatic prostate cancer. Nat. Cancer 1, 1041–1053 (2020).
Watson, P. A., Arora, V. K. & Sawyers, C. L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711 (2015).
Robinson, D. et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015).
Beltran, H. et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 22, 298–305 (2016).
Davies, A. H., Beltran, H. & Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 15, 271–286 (2018).
Bluemn, E. G. et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 32, 474–489 e476 (2017).
Aggarwal, R. et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol. 36, 2492–2503 (2018).
Fujii, M., Sekine, S. & Sato, T. Decoding the basis of histological variation in human cancer. Nat. Rev. Cancer 24, 141–158 (2024).
Beltran, H. et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 25, 6916–6924 (2019).
Ku, S. Y. et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017).
Dardenne, E. et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 30, 563–577 (2016).
Mu, P. et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88 (2017).
Bishop, J. L. et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 7, 54–71 (2017).
Han, M. et al. FOXA2 drives lineage plasticity and KIT pathway activation in neuroendocrine prostate cancer. Cancer Cell 40, 1306–1323 e1308 (2022).
Svensson, C. et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 42, 999–1015 (2014).
Reina-Campos, M. et al. Increased serine and one carbon pathway metabolism by PKCl/i deficiency promotes neuroendocrine prostate cancer. Cancer Cell 35, 1–16 (2019).
Reina-Campos, M., Diaz-Meco, M. T., Moscat, J. The complexity of the serine glycine one-carbon pathway in cancer. J. Cell Biol. 219, e201907022 (2020).
Blackledge, N. P. & Klose, R. J. The molecular principles of gene regulation by Polycomb repressive complexes. Nat. Rev. Mol. Cell Biol. 22, 815–833 (2021).
Kim, K. H. & Roberts, C. W. Targeting EZH2 in cancer. Nat. Med 22, 128–134 (2016).
Puca, L. et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 9, 2404 (2018).
Xu, K. et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469 (2012).
Davies, A. et al. An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer. Nat. Cell Biol. 23, 1023–1034 (2021).
Kim, J. et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25, 2808–2820 e2804 (2018).
Abida, W. et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 116, 11428–11436 (2019).
Brady, N. J. et al. Temporal evolution of cellular heterogeneity during the progression to advanced AR-negative prostate cancer. Nat. Commun. 12, 3372 (2021).
Gao, D. et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187 (2014).
Rusilowicz-Jones, E. V., Urbe, S. & Clague, M. J. Protein degradation on the global scale. Mol. Cell 82, 1414–1423 (2022).
Cha, T. L. et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310, 306–310 (2005).
Xue, Y. et al. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell Proteom. 7, 1598–1608 (2008).
Nikitina, A. S. et al. Novel RNA biomarkers of prostate cancer revealed by RNA-seq analysis of formalin-fixed samples obtained from Russian patients. Oncotarget 8, 32990–33001 (2017).
Ross-Adams, H. et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: a discovery and validation cohort study. EBioMedicine 2, 1133–1144 (2015).
Shan, M. et al. Molecular analyses of prostate tumors for diagnosis of malignancy on fine-needle aspiration biopsies. Oncotarget 8, 104761–104771 (2017).
Zhong, W. D. et al. SOXs in human prostate cancer: implication as progression and prognosis factors. BMC Cancer 12, 248 (2012).
Stoss, O. et al. Transcriptional profiling of transurethral resection samples provides insight into molecular mechanisms of hormone refractory prostate cancer. Prostate Cancer Prostatic Dis. 11, 166–172 (2008).
Yun, S. J. et al. Transcriptomic features of primary prostate cancer and their prognostic relevance to castration-resistant prostate cancer. Oncotarget 8, 114845–114855 (2017).
Hsieh, A. C. et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17, 249–261 (2010).
Pourdehnad, M., Truitt, M. L., Siddiqi, I. N., Ducker, G. S. & Shokat, K. M. Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc. Natl. Acad. Sci. USA 110, 11988–11993 (2013).
Jiang, C. et al. Ring domains are essential for GATOR2-dependent mTORC1 activation. Mol. Cell 83, 74–89 e79 (2023).
Reich, S. H. et al. Structure-based design of pyridone-aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) inhibition. J. Med. Chem. 61, 3516–3540 (2018).
Fresno, M., Jimenez, A. & Vazquez, D. Inhibition of translation in eukaryotic systems by harringtonine. Eur. J. Biochem. 72, 323–330 (1977).
Hsieh, A. C. et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 (2012).
Lee, A. S., Kranzusch, P. J. & Cate, J. H. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114 (2015).
Lamper, A. M., Fleming, R. H., Ladd, K. M. & Lee, A. S. Y. A phosphorylation-regulated eIF3d translation switch mediates cellular adaptation to metabolic stress. Science 370, 853–856 (2020).
Davies, A., Zoubeidi, A., Beltran, H. & Selth, L. A. The transcriptional and epigenetic landscape of cancer cell lineage plasticity. Cancer Discov. 13, 1771–1788 (2023).
Giafaglione, J. M. et al. Prostate lineage-specific metabolism governs luminal differentiation and response to antiandrogen treatment. Nat. Cell Biol. 25, 1821–1832 (2023).
Park, S. H. et al. Going beyond Polycomb: EZH2 functions in prostate cancer. Oncogene 40, 5788–5798 (2021).
Lu, W. et al. SKP2 loss destabilizes EZH2 by promoting TRAF6-mediated ubiquitination to suppress prostate cancer. Oncogene 36, 1364–1373 (2017).
Chen, S. et al. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat. Cell Biol. 12, 1108–1114 (2010).
Nouruzi, S. et al. ASCL1 activates neuronal stem cell-like lineage programming through remodeling of the chromatin landscape in prostate cancer. Nat. Commun. 13, 2282 (2022).
Jana, S. et al. Transcriptional-translational conflict is a barrier to cellular transformation and cancer progression. Cancer Cell 41, 853–870.e813 (2023).
Kovalski, J. R., Kuzuoglu-Ozturk, D. & Ruggero, D. Protein synthesis control in cancer: selectivity and therapeutic targeting. EMBO J. 41, e109823 (2022).
Furic, L. et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc. Natl. Acad. Sci. USA 107, 14134–14139 (2010).
Brina, D. et al. The Akt/mTOR and MNK/eIF4E pathways rewire the prostate cancer translatome to secrete HGF, SPP1 and BGN and recruit suppressive myeloid cells. Nat. Cancer 4, 1102–1121 (2023).
He, M. X. et al. Transcriptional mediators of treatment resistance in lethal prostate cancer. Nat. Med. 27, 426–433 (2021).
Linares, J. F. et al. PKClambda/iota inhibition activates an ULK2-mediated interferon response to repress tumorigenesis. Mol. Cell, 81, 4509–4526.e10 (2021).
Ma, L. et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell 152, 599–611 (2013).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Han, C. et al. Polysome profiling followed by quantitative PCR for identifying potential micropeptide encoding long non-coding RNAs in suspension cell lines. STAR Protoc. 3, 101037 (2022).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Danecek P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Xiao, Z., Zou, Q., Liu, Y. & Yang, X. Genome-wide assessment of differential translations with ribosome profiling data. Nat. Commun. 7, 11194 (2016).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021).
Acknowledgements
Research was supported by grants from the National Cancer Institute of the National Institutes of Health under award numbers R01CA246765 to M.T.D.-M. and H.B.; R01CA277857 to M.T.D.-M; R01CA265892, R01CA250025, and R01CA275846 to J.M.; R50CA283476 to J.F.L. R50CA265332 to A.D.; R01CA230913 to D.S.R.; P50CA211024 to D.S.R., H.B., F.K., B.D.R., and J.M.M; R37CA230617, R01CA276308, and R01GM135362 to A.C.H. We thank Tavonna Bryant and the personnel of the Translational Research Program in the Department of Pathology and Laboratory Medicine, Genomics, the Scientific Computing Unit, the Proteomics and Metabolomics Core Facility, the Optical Microscopy Cores at Weill Cornell Medicine, the Englander Institute of Precision Medicine, and Eric Rosiek of the Molecular Cytology Core at Memorial Sloan Kettering Cancer Center, for technical assistance. M.T.D.-M. and J.M. are Homer T. Hirst III Professors of Oncology in Pathology.
Author information
Authors and Affiliations
Contributions
Conceptualization, M.T.D-M., J.M., S.S.C., J.F.L., T.C.D., and A.D.; Methodology, M.T.D-M., J.M., S.S.C., J.F.L., T.C.D., A.D. and A.M.; Investigation, S.S.C., J.F.L.,T.C.D., A.D., M.I.K.K., M.O., N.J.B., M.R-C., V.B.V., M.K.B., and F.K.; Resources, J.M.M., B.D.R., J.M., O.E., A.C.H., D.W.G., D.S.R., and H.B.; Writing–Original Draft, M.T.D.-M., J.M., J.F.L., A.D.,T.C.D., and S.S.C.; Writing–Review & Editing, all authors; Supervision, M.T.D-M., and J.M., and Funding Acquisition, M.T.D-M., and J.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chatterjee, S.S., Linares, J.F., Cid-Diaz, T. et al. Increased translation driven by non-canonical EZH2 creates a synthetic vulnerability in enzalutamide-resistant prostate cancer. Nat Commun 15, 9755 (2024). https://doi.org/10.1038/s41467-024-53874-2
Received
Accepted
Published
DOIhttps://doi.org/10.1038/s41467-024-53874-2



Comments
Post a Comment